Publicado el 1 de julio de 2008 | http://doi.org/10.5867/medwave.2008.06.502
Inflamación, daño y reparación en enfermedades reumáticas
Inflammation, damage and repair in rheumatic diseases
Resumen
Este texto completo es una transcripción editada y revisada de una conferencia dictada en el Curso Actualizaciones en Dolor e Inflamación en Reumatología, organizado por la Sociedad Chilena de Reumatología, durante los días 5 y 6 de Octubre de 2007, cuyo director fue el Dr. Carlos Fuentealba.
Inflamación: definición
La inflamación es un proceso complejo e inespecífico, que se caracteriza por modificaciones locales y coordinadas de los vasos sanguíneos y el tejido conectivo; se relaciona con el proceso de reparación, que consiste en la regeneración de las células parenquimatosas dañadas, y con la cicatrización, que se caracteriza por la proliferación de tejido fibroblástico. La inflamación es un proceso propio del tejido conectivo vascularizado y en ella participan el plasma y las células circulantes y residentes del tejido conectivo. Se caracteriza clínicamente por calor, rubor, tumor, dolor y pérdida de función. Los elementos principales que participan en este proceso son los mediadores de la inflamación, vasos sanguíneos y varios tipos de células: granulocitos, monocito-macrófagos, plaquetas, células endoteliales y fibroblastos, estos últimos con un importante rol en la inflamación crónica (Fig. 1).
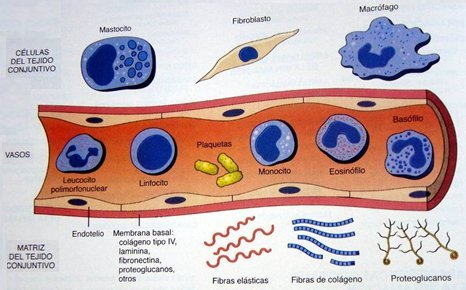 Tamaño completo
Tamaño completo En forma resumida, el proceso inflamatorio se inicia cuando los leucocitos que viajan por el centro del torrente sanguíneo se adhieren al endotelio, atraídos por moléculas que produce el propio endotelio y otras células inflamatorias; luego el leucocito migra entre las células endoteliales y llega al foco inflamatorio, donde producirá la destrucción tisular. Entre las principales características de la inflamación se encuentra el aumento del flujo sanguíneo, el aumento de la permeabilidad capilar y venular y la movilización de células inflamatorias hacia el foco en que se produjo el daño.
Mediadores de la inflamación
Los mediadores de la inflamación pueden ser de origen plasmático o celular. Entre los mediadores de origen plasmático están: el sistema de complemento; el sistema de coagulación, que termina con la producción de trombina; el sistema fibrinolítico, que produce fibrinopéptidos y el sistema de quininas, que genera bradiquinina. Como se observa en la figura 2, cuando se produce daño endotelial se activa inicialmente el sistema de complemento, el que induce una activación simultánea de la cascada de la coagulación, del sistema fibrinolítico y de la vía de la bradiquinina. La activación de estos sistemas conduce a un aumento de la permeabilidad vascular y vasodilatación, entre otros efectos.
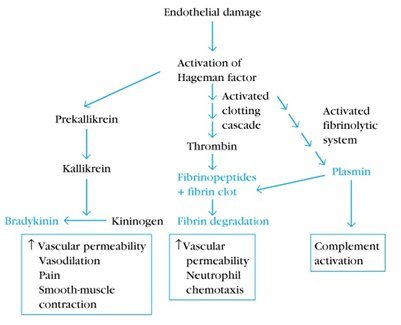 Tamaño completo
Tamaño completo El sistema del complemento produce aumento de la permeabilidad vascular, lo que permite la extravasación de plasma, inmunoglobulinas y células que participarán en la inflamación, como linfocitos y polimorfonucleares; esto se logra principalmente gracias a la acción de los componentes C3a, C4a y C5a, mientras que el fragmento C3b participa en la opsonización de patógenos y remoción de complejos inmunes. El sistema del complemento también tiene un rol en la liberación de citoquinas y en el reclutamiento celular, lo que determina un medio proinflamatorio. Este sistema es muy relevante en reumatología, ya que muchas enfermedades autoinmunes del tejido conectivo, como la artritis reumatoide (AR), el lupus eritematoso sistémico (LES) y la enfermedad de Sjögren son mediadas por complejos antígeno-anticuerpo que activan al sistema del complemento (1).
Entre los mediadores de origen celular destacan histamina, serotonina y heparina, que actúan en la fase inicial del proceso inflamatorio, además de prostaglandina E2, leucotrieno B4 y factor activador plaquetario (PAF). Dentro de los mediadores celulares el grupo de las citoquinas juega un rol fundamental en la propagación y cronicidad de la inflamación, en especial la interleuquina 1 (IL-1) y el factor de necrosis tumoral alfa (TNF alfa). Otras citoquinas importantes en este proceso son la IL-8 y la IL-17.
El macrófago es el principal productor de citoquinas en la inflamación crónica; cuando es activado por distintas señales de peligro, como agentes patógenos, complejos inmunes y otras citoquinas, esta célula libera IL-1, que activa a endotelio y linfocitos; además produce TNF alfa, que también actúa sobre el endotelio, donde induce reclutamiento celular, favorece la expresión de moléculas de adhesión y estimula la extravasación de plasma. Otras citoquinas importantes en la inflamación aguda y que son sintetizadas por los macrófagos son IL-8, IL-6 e IL-l2. Existen dos grandes vías de activación de los macrófagos: una dependiente de citoquinas como TNF alfa y otra independiente de citoquinas, que se desencadena frente al contacto con linfocitos T, con proteínas denaturadas de la matriz celular o con algunas hormonas (2).
En la inflamación crónica, así como en algunos modelos experimentales de AR, la IL-1 favorece la destrucción tisular, aumentando la expresión de metaloproteinasas. Sin embargo, su rol patogénico más importante en la inflamación crónica consiste en dificultar la reparación de tejidos, al inhibir la síntesis de colágeno y proteoglicanos. En cambio, TNF alfa es una citoquina más bien proinflamatoria y su acción principal no es impedir la reparación (Fig. 3).
 Tamaño completo
Tamaño completo La IL-17, descubierta más recientemente, es una citoquina producida por una subpoblación de linfocitos T CD4 conocidos como linfocitos Th17. Esta interleuquina tiene un rol sinérgico con IL-1 y TNF alfa, estimulando la producción de citoquinas proinflamatorias y quemoquinas por parte del fibroblasto y del macrófago sinoviales.
Rol del endotelio en la inflamación
La célula endotelial produce una serie de mediadores con función biológica, entre ellos el óxido nítrico (NO), las ceramidas y la endotelina 1 (ET-1). La producción intermitente de NO favorece el daño tisular, pero si se sintetiza de manera sostenida y continua juega un rol protector contra el daño, al inhibir la producción de quimioquinas e impedir el reclutamiento celular. De hecho, el efecto antiinflamatorio del uso crónico de acido acetilsalicílico (AAS) en la microvasculatura se debe a la inhibición de la interacción entre leucocito y célula endotelial, debido a un aumento en la producción de NO mediado por la enzima 15 epilipooxigenasa A4. El NO tiene además la capacidad de inhibir la exocitosis de ceramidas por parte de los gránulos de las células endoteliales. Estos gránulos, llamados cuerpos de Weibel-Palade, contienen sustancias procoagulantes y proinflamatorias, como Factor de von Willebrand, selectina E, selectina P y ET-1.
Mecanismos de migración leucocitaria
La movilización de los leucocitos hacia el foco inflamatorio se realiza mediante un conjunto de procesos seriados: marginación, adhesión, diapedesis y quimiotaxis. Normalmente los leucocitos se ubican en el centro del vaso sanguíneo, pero cuando se inicia el proceso inflamatorio se marginan y luego, por la acción de distintas moléculas de adhesión, ruedan sobre la pared endotelial, en lo que se denomina la etapa de adhesión laxa. En ese momento los leucocitos se activan y expresan otras moléculas de adhesión, que determinan una firme unión con el endotelio, dando paso a la etapa de adhesión firme, que está mediada por el aumento de la afinidad entre las integrinas leucocitarias y sus receptores endoteliales. La etapa de adhesión firme es transitoria, el leucocito pronto se “despega” del endotelio y atraviesa el vaso entre las células endoteliales, fenómeno conocido como diapedesis, atraído por factores quimiotácticos, principalmente las quimioquinas producidas por las células que se encuentran infiltrando el tejido dañado. En la figura 4 se observa el rol de distintas moléculas de adhesión en la sinovitis: los leucocitos se adhieren al endotelio por acción de las distintas moléculas de adhesión para luego migrar hacia la sinovial, con la consiguiente formación de pannus.
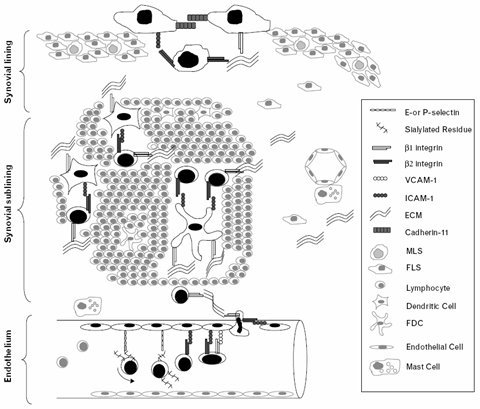 Tamaño completo
Tamaño completo La fagocitosis en la inflamación aguda está mediada predominantemente por los polimorfonucleares (PMN) neutrófilos, mientras que en la inflamación crónica es llevada a cabo por monocito-macrófagos. Para ello, primero el leucocito reconoce al patógeno y lo engloba y endocita, formando un fagolisosoma en su interior. Este fagolisosoma contiene gránulos con distintas moléculas, que destruirán al patógeno. El contenido de los gránulos varía según se trate de un PMN neutrófilo o de un monocito-macrófago: en el primero contienen enzimas hidrolíticas, mieloperoxidasas (MPO), proteínas catiónicas y lisozimas; los gránulos del monocito-macrófago contienen enzimas lisozomales, hidrolasas ácidas, proteasas neutras, proteínas catiónicas y fosfolipasa A2.
Factores involucrados en el paso de inflamación aguda a crónica
La inflamación aguda puede evolucionar de distintas formas:
- Resolución completa.
- Formación de abscesos.
- Curación con destrucción tisular, formación de fibrina y sustitución por tejido conectivo.
- Progresión hacia inflamación crónica, dependiendo de la persistencia del agente causal y de factores propios del hospedero.
En la figura 5 se observa un modelo de persistencia de inflamación, en la cual el fibroblasto tiene un rol muy importante. Durante la inflamación aguda se producen señales de peligro, que pueden ser citoquinas, quimioquinas o bacterias que van a activar tanto al fibroblasto como al macrófago, para que éstos produzcan una serie de citoquinas y quimioquinas proinflamatorias. Estas señales de peligro van a activar también a células dendríticas inmaduras, que migrarán hacia los linfonodos regionales para iniciar una respuesta inmune adaptativa. El paso de una respuesta inmune innata o inespecífica a una de tipo adaptativa es crucial en el proceso de inflamación crónica. En el mismo modelo, los linfocitos, macrófagos y fibroblastos infiltran el tejido y persisten en él por mucho tiempo, prolongando el proceso inflamatorio.
 Tamaño completo
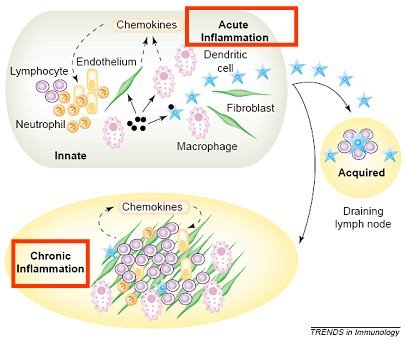
Tamaño completo Se denomina inflamación crónica a la que presenta un curso prolongado, de semanas a meses, con signos de inflamación aguda, destrucción tisular y reparación. Su inicio puede ser solapado y asintomático. Desde el punto de vista microscópico, se caracteriza por infiltración de células mononucleares (linfocitos y macrófagos), evidencias de destrucción tisular provocada por estas células e intentos de reparación, mediante angiogénesis y fibrosis. El macrófago tisular es la célula dominante en la inflamación crónica. Durante las primeras 48 horas que siguen al daño tisular, el monocito migra hacia el extravascular, donde se diferencia a macrófago tisular y se localiza en el foco inflamatorio, adoptando distintos nombres según el tejido en cual se sitúe. En la inflamación crónica, la acumulación de macrófagos persiste por distintos mecanismos, entre ellos, reclutamiento continuo desde la circulación y proliferación local de los macrófagos; recientemente se ha propuesto que estas células tendrían una sobrevida prolongada en el foco inflamatorio. Los fibroblastos jugarían un rol importante en la mantención de estos fenómenos (3).
En la figura 6 se ilustra la diferencia que hay entre inflamación aguda y crónica en cuanto a la renovación del pool celular leucocitario. El balance dinámico de la acumulación celular, en cualquier tejido, depende del balance entre reclutamiento celular, división, migración y muerte. En la inflamación aguda normal esta homeostasis se mantiene, permitiendo la resolución de la inflamación, mientras que en la inflamación crónica se produce una acumulación inapropiada de leucocitos, especialmente de macrófagos, debido a una producción también inapropiada de elementos que favorecen la retención y la supervivencia, por parte de los fibroblastos. Así, estas células aumentan la producción de interferón beta, que inhibe la muerte celular, prolongando la sobrevida de linfocitos y macrófagos y producen SDF-1 (stromal cell-derived factor-1), que inhibe la migración de los leucocitos.
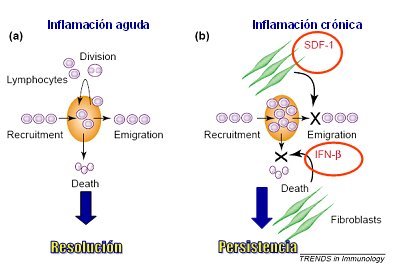 Tamaño completo
Tamaño completo Así, durante la inflamación crónica los macrófagos eliminan al agente que está causando el daño y dan inicio a la reparación, pero al mismo tiempo son responsables de gran parte de la lesión tisular circundante. Una vez que son activados por los mediadores que ya se mencionaron, los macrófagos producen daño tisular a través de cuatro mecanismos principales: producción de especies reactivas de oxígeno (ROS), inducción de síntesis de metaloproteasas, activación de factores de coagulación y por último, producción de metabolitos de NO y ácido araquidónico. Por otro lado, en el foco inflamatorio crónico los macrófagos participan en la génesis de fibrosis, a través de la producción de factores de crecimiento involucrados en la proliferación de fibroblastos (PDGF, TGFb, FGF), producción de factores angiogénicos (FGF, VEGF) y estimulando el depósito de colágeno a través de IL-13 y TGFb (Figura 7).
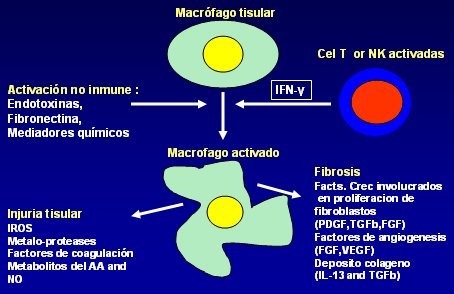 Tamaño completo
Tamaño completo
Reparación y fibrosis
En el proceso de reparación de tejido conectivo o fibrosis, se distinguen cuatro etapas:
- Angiogénesis.
- Formación de tejido de granulación, por migración y proliferación de fibroblastos.
- Depósito de matriz extracelular (colágeno, elastina, etc.)
- Remodelación u organización de tejido fibroso.
La angiogénesis o desarrollo de nuevos vasos sanguíneos es un proceso normal, necesario para la reparación tisular y el restablecimiento del flujo sanguíneo luego de una lesión. Corresponde a un proceso altamente controlado mediante quimioquinas inhibidoras o angioestáticas, versus favorecedoras o proangiogénicas; sin embargo, en muchas enfermedades el organismo pierde el control sobre este proceso y, como consecuencia, se establecen estados de enfermedad dados por exceso o déficit de angiogénesis. Entre las enfermedades reumatológicas que cursan con exceso de angiogénesis están la AR y la psoriasis, mientras que en la esclerodermia existe una angiogénesis deficiente. Esta diferencia se debe a algunas quimioquinas de la familia CXC, que tienen un rol fundamental en la inflamación crónica y la angiogénesis (4). En la figura 8 se muestran las quimioquinas CXC con efecto proangiogénico y aquellas con efecto angioestático, con sus respectivos receptores de membrana.
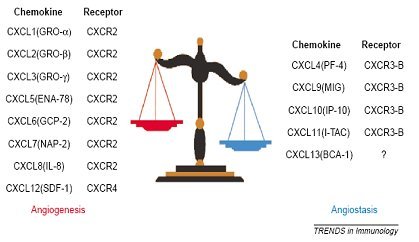 Tamaño completo
Tamaño completo El proceso de angiogénesis y neovascularización es crítico en inflamación crónica y fibrosis. Una vez que se produce la lesión y se desarrolla un trombo las plaquetas comienzan a producir quimioquinas, de las cuales, CXCL4, CXCL5 y CXCL7 tienen como función reclutar neutrófilos hacia el foco inflamatorio agudo, mientras que CXCL1 y CXCL8 favorecen la angiogénesis y la formación de tejido granulatorio; posteriormente el epitelio dañado mantiene la producción de CXCL8, favoreciendo la reparación del mismo. Por otro lado, las quimioquinas CXCL9, CXCL10 y CXCL11 inhiben la inflamación e inmovilizan a los fibroblastos, lo que constituye una forma de regulación del proceso.
Frente al daño endotelial se activa el sistema inmune innato, comenzando por la activación de los macrófagos; posteriormente se activa el sistema inmune adaptativo, lo que lleva a la producción de citoquinas, sobre todo aquellas de tipo Th2 (IL4, IL5, IL8, IL13), que activan al fibroblasto y favorecen la angiogénesis. Entre las células que promueven la proliferación de los fibroblastos están los mastocitos, metiante la producción de histamina, serotonina y heparina y los eosinófilos, macrófagos, células epiteliales y los propios fibroblastos (figura 9).
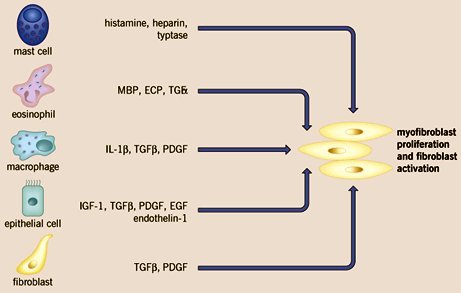 Tamaño completo
Tamaño completo El fibroblasto activado produce una serie de sustancias proinflamatorias, como quimioquinas, citoquinas, factores de crecimiento, metaloproteinasas e inhibidores tisulares de metaloproteinasas, entre otros (figura 10).
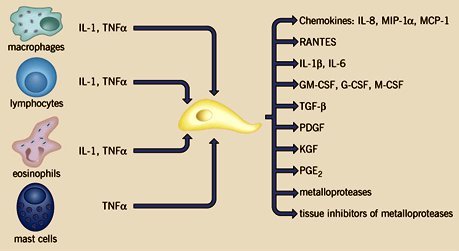 Tamaño completo
Tamaño completo También produce componentes de la matriz extracelular que llevan a la reparación, como colágeno, elastina, fibronectina y proteoglicanos (figura 11).
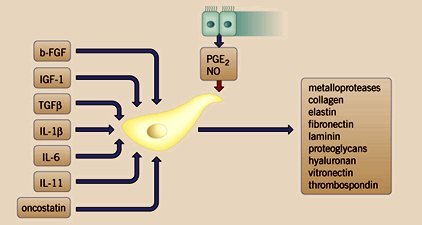 Tamaño completo
Tamaño completo En el caso de la esclerodermia, un modelo publicado en 2005 plantea la siguiente hipótesis para explicar el mecanismo de la fibrogénesis: existe un progenitor mesenquimático circulante, que se transforma en fibroblasto activo por acción de ET-1 y, posteriormente, se diferencia a miofibroblasto por acción de mediadores de inflamación locales. Este miofibroblasto produce gran cantidad de componentes de la matriz extracelular, favoreciendo la formación de cicatrices, fibrosis y contracción del tejido; a diferencia del fibroblasto común, el miofibroblasto tiene la capacidad de contraerse (5). Este tipo de célula infiltra los tejidos y es responsable de la atrofia que ocurre en algunas enfermedades reumatológicas crónicas (figura 12).
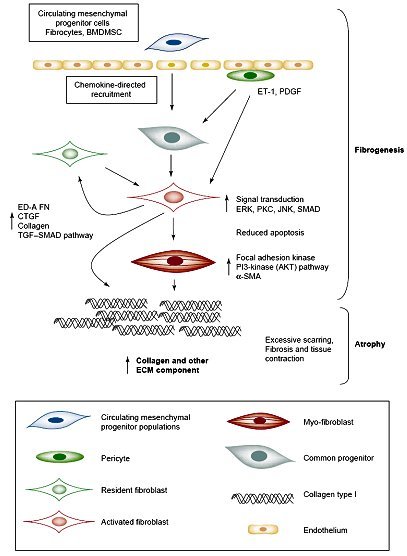 Tamaño completo
Tamaño completo
Fibrosis
- La fibrosis es el resultado final de la inflamación crónica y la reparación.
- Consiste en una acumulación excesiva de componentes de la matriz extracelular, especialmente el colágeno.
- Los macrófagos y fibroblastos son los principales efectores involucrados en la patogenia de la fibrosis.
- Existen mediadores profibróticos, como IL-13 y TGF beta (Transforming growth factor beta), capaces de amplificar el proceso; el primero, producido por linfocitos T, favorece la migración de fibroblastos y la proliferación del miofibroblasto, mientras que TGF beta es producido por el endotelio dañado y actúa activando a los macrófagos.
- La degradación del colágeno es controlada por metaloproteinasas de la matriz, activadas por IFN gamma.
- El incremento neto de colágeno tisular es controlado por un balance entre mecanismos opuestos.
- En el caso de la fibrosis por daño crónico, la respuesta inmune adaptativa tiene un rol muy importante.
A modo de ejemplo, en la fibrosis pulmonar asociada a enfermedades reumáticas existiría un predominio de los factores activadores de la cascada de coagulación y una disminución de los factores fibrinolíticos; por lo tanto, estos elementos entonces deberían ser regulados para evitar las manifestaciones de la enfermedad.
