Publicado el 1 de mayo de 2004 | http://doi.org/10.5867/medwave.2004.04.3488
Linfoma de células del manto
Mantle cell lymphoma
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso Teórico y Seminarios de Oncología Básica, organizado por el Centro de Oncología Preventiva de la U. de Chile entre el 2 de abril y 7 de octubre de 2003. Director: Dr. José Manuel Ojeda.
En la década de los 70, algunos investigadores de los Estados Unidos describieron el linfoma centrocítico, caracterizado por la presencia de células hendidas de núcleo irregular, que podía clasificarse dentro de los linfomas no Hodgkin (LNH). En los cortes de ganglios linfáticos se observó que los tumores tenían un patrón de crecimiento difuso y estaban compuestos de una mezcla de células linfoides pequeñas, algunas con núcleo redondo, como el linfoma linfocítico, y otras con núcleo hendido, como el linfoma de células pequeñas hendidas. Los estudios inmunológicos detectaron una positividad moderada a intensa para Ig de superficie.
Recién en 1982, Weisenburger, Raffeld y Jaffe describieron un subtipo de linfoma folicular que se caracterizaba por la proliferación atípica de células B pequeñas dispuestas en mantos anchos alrededor de centros germinales benignos, para cuya denominación propusieron el término de linfoma de la zona del manto. Los estudios clínicos, moleculares y de inmunofenotipo permitieron establecer que se trataba de una variante morfológica de una familia de linfomas que actualmente se conoce como linfomas de células del manto (LCM).
Las células neoplásicas del LCM parecen corresponder a linfocitos B naive que normalmente residen en los folículos linfoides primarios y en las zonas del manto de los folículos secundarios. Como tales, corresponden a una alteración de las células B foliculares, que al transformarse en respuesta a un antígeno, se convierten en células del centro germinal.
La relación entre las formas nodular y difusa del LCM es biológicamente análoga a la del linfoma de células del centro germinal de tipo folicular y difuso respectivamente, mientras que la forma blástica es análoga a un linfoma transformado.
Histología
Citológicamente, el LCM está formado por una población homogénea de células linfoides de pequeño a mediano tamaño, con núcleo irregular, cromatina moderadamente condensada y citoplasma escaso; sin embargo, a veces las células no cumplen estas características, pudiendo mostrar un amplio espectro de variaciones, tanto en el núcleo como en el citoplasma. Existe una atipia celular moderada, con células que muestran individualmente menos irregularidad en los núcleos que lo que se ve típicamente en los linfomas foliculares y difusos de células pequeñas hendidas.
Weisenburger describió dos patrones histológicos: el patrón de zona del manto y el patrón difuso, que parecían tener implicancia pronóstica. Fisher (1), en 1995, describió la variante difusa blástica, caracterizada por su mal resultado, ya que la sobrevida observada era menor de 2 años, en contraste con la variante nodular, que tenía una sobrevida libre de enfermedad de 5 años. Sin embargo, incluyó como variantes nodulares casos con configuración de zona del manto.
En el 20% de los LCM las células son más grandes que lo habitual y tienen la cromatina finamente dispersa y un nucléolo pequeño; estas células se han descrito en la variante blástica del LCM (2). El índice mitótico generalmente es bajo en las formas linfocíticas, pero está aumentado en la variante blástica.
El nombre de este linfoma se debe a su patrón de crecimiento peculiar, rodeando los centros germinales preexistentes y sustituyendo los mantos foliculares normales. Sin embargo, este patrón característico es poco frecuente, debido al crecimiento progresivo, que tiende a colapsar los centros germinales y suele presentarse como un patrón nodular difícil de distinguir del linfoma folicular, para, finalmente, infiltrar en forma difusa. En algunos casos, algunos nódulos neoplásicos sin centro germinal podrían simular folículos primarios.
En la figura 1 se muestra una serie de imágenes que ilustran distintas formas de presentación de un LCM.
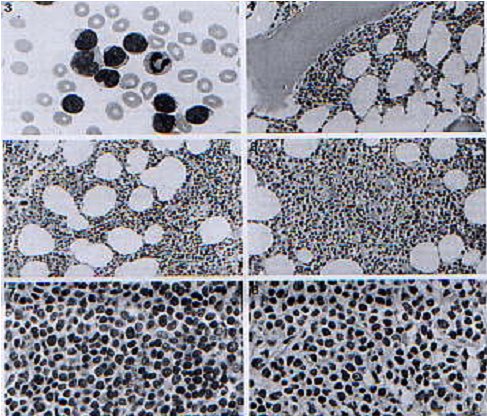 Tamaño completo
Tamaño completo Figura 1. Histología en distintos patrones de presentación del linfoma de células del manto.
(British Journal of Haematology 101:305,1998).
Fotografía 3. Frotis de sangre periférica en LCM
Fotografía 4. Patrón paratrabecular de compromiso médula ósea
Fotografía 5. Patrón intersticial de compromiso médula ósea
Fotografía 6. Patrón nodular de compromiso médula ósea
Fotografía 7. Variedad blástica de LCM en biopsia de médula ósea.
Fotografía 8. Variante linfomonocitoide de LCM en biopsia de médula ósea.
Características clínicas
El LCM constituye un 2,5 a 4% de todos los LNH, en los Estados Unidos, alcanzando cifras de 7 a 9% en Europa (2). La Working Formulation divide a los LNH en subtipos de bajo, intermedio y alto grado de acuerdo con las correspondientes sobrevidas; en cambio, Kiel usa las categorías de bajo o alto grado según las características citológicas, independientemente de la sobrevida.
El LCM no aparece como una entidad definida en la Working Formulation, porque en ese momento aún no se tipificaba, y probablemente se le incluyó en el grupo del linfoma difuso de células pequeñas y hendidas; sin embargo, en la clasificación Kiel es considerado como un linfoma de bajo grado.
Los pacientes con LCM tienen en promedio 60 años en el momento del diagnóstico, con un claro predominio en el sexo masculino. Generalmente se presentan en etapas avanzadas, (III/IV), con linfadenopatía generalizada y compromiso hepático y de médula ósea, pero menos de la mitad tiene síntomas sistémicos. La esplenomegalia está presente en 60% de los pacientes en el momento del diagnóstico; en el tipo nodular esto llega al 80%, y puede ser considerable.
La mayoría de los pacientes con LCM que tienen una enfermedad nodal de base, tienen escaso compromiso de sangre periférica. La anemia moderada no es infrecuente, mientras que la trombocitopenia se ve en menos de 15% de los pacientes. En un estudio, más del 77% de los casos tenía evidencias morfológicas en sangre periférica, con un bajo número de células de linfoma circulando (3). En 20 a 40% de los casos (2) existe una linfocitosis periférica mayor de 4.000/ul, siendo raros los valores mayores a 20.000.
Las células neoplásicas en sangre y médula ósea de un paciente pueden ser bastante heterogéneas en apariencia; los estudios inmunológicos por citometría de flujo pueden ser útiles para el diagnóstico de tales poblaciones. La hipogamaglobulinemia, la gamapatía monoclonal y el Coombs positivo también son poco frecuentes, y los frotis de sangre y médula ósea reflejan la población linfoide presente en los linfonodos.
Compromiso extranodal
La infiltración de otros órganos por el LCM no es rara, debido a que el diagnóstico generalmente se hace en etapas avanzadas. El bazo está aumentado de tamaño, particularmente en el patrón de tipo nodular, y al microscopio se puede ver que la pulpa blanca tiene áreas marcadamente aumentadas, por proliferación de células linfoides atípicas y centros germinales aparentemente reactivos. El compromiso hepático es común, y se caracteriza por la infiltración linfoide portal. Otros sitios de compromiso extranodal son el tracto gastrointestinal y el anillo de Waldeyer (20-30% de los casos). La poliposis linfomatosa múltiple intestinal puede sugerir el diagnóstico de LCM.
El compromiso extranodal es independiente del tipo histológico, pero la infiltración de la médula ósea se presenta en 90% de los pacientes con variedad de crecimiento difuso y sólo en 33% de los casos con pattern de zona del manto (4). El compromiso de médula ósea puede ser focal, intersticial, difuso o paratrabecular.
Características inmunológicas
En cortes congelados, las células del LCM expresan un fenotipo característico, con antígenos de estirpe B como CD 20 y CD 79, y al igual que sus equivalentes normales, también expresan Ig M y D de superficie. En más del 90% de los casos expresan CD5 y CD43 (anómalo), que están presentes en los linfocitos T.
La relación kappa/lambda está invertida en el LCM; aproximadamente 60% de los casos expresan cadenas livianas lambda monoclonales. Las células neoplásicas son positivas para una serie de marcadores B (CD19, 20, 22 y 24) y antígeno HLA-DR; en cambio, son negativas para antígeno CD10 (CALLA) y CD23, lo que lo distingue del linfoma folicular CD10. Los casos de variante blástica son menos propensos a expresar IgD, CD5 y CD43, e incluso pueden expresar antígeno CD10.
La inmunohistoquímica puede ser útil para distinguir un proceso reactivo de un LCM, por la monoclonalidad y la positividad para CD5 y CD43 propios de las células neoplásicas, que permiten distinguir claramente las células del LCM de la hiperplasia folicular, la hiperplasia de la zona del manto y la hiperplasia angiofolicular. Esta regla también es válida para distinguir el LCM difuso de proliferaciones linfoides reactivas de células estirpe B.
Citogenética
El LCM es un tipo especial de linfoma no Hodgkin, que se caracteriza por la presencia de la translocación cromosómica balanceada t(11;14)(q13;q32). Esta anormalidad yuxtapone el gen CCND1 (11q13) con el gen de la IgH (14q32), lo que resulta en una sobreexpresión del gen PRAD1, que codifica la ciclina D1. La detección de la expresión nuclear de ciclina D1 es el rasgo más característico de esta entidad, y se considera casi exclusiva de este tipo de linfoma (por el momento).
Como consecuencia de la desrregulación del ciclo celular se encuentra un elevado índice mitótico(>2,5) medible por la expresión de Ki 67.
Se ha visto que el LCM a menudo se asocia a otras anormalidades cariotípicas adicionales, y la mayoría de los estudios demuestra que la del (13)(q14) es la más frecuente, encontrándose en 30 a 70% de los casos (5). Otras anormalidades comunicadas involucran a los cromosomas 1, 3, 6, 9 y 17, pero no se ha definido el significado clínico de éstas.
Wlodarska y cols (6) encontraron que 94% de los casos de LCM tenía anormalidades cariotípicas adicionales a la t(11;14), lo que concordaba con los resultados de estudios previos, usando citogenética convencional. Concluyeron que la frecuente presencia de anormalidades cariotípicas adicionales sugiere su importancia en la patogenia del LCM, lo que concuerda con estudios previos realizados en ratones transgénicos, ya que la sobreexpresión de ciclina D1 es insuficiente, por sí misma, como causa de oncogénesis.
Las anormalidades del cromosoma 17, 21 y 22 se relacionan con la presentación leucémica; por otro lado, el LCM nodal adquiere anormalidades cariotípicas adicionales en la medida que progresa a una fase leucémica de la enfermedad. Aparte de la t(11;14), las anormalidades del cromosoma 13 son muy frecuentes en el LCM; la del(13)(q14) se ha encontrado también en el linfoma folicular leucémico y en algunos casos agresivos de mieloma múltiple.
Características moleculares
La t(11;14) involucra un error en la unión V-D-J durante el reordenamiento génico de la cadena pesada de las Ig, resultando en el movimiento del oncogen adyacente a BCL1 (11q13) hacia la proximidad de la región enhancer del gen de la cadena pesada de las Ig (14q32).
Los quiebres en la región posterior ocurren precozmente en el desarrollo de los linfocitos B, siendo mediados por el sistema recombinasa, mientras que el sitio 11q13 aparece como un sitio común frágil. Los puntos de quiebre en el locus BCL1 no están bien estudiados, aunque el 30 a 40% de los casos de LCM tiene puntos de quiebre en la región del cluster de translocación mayor.
El oncogen desrregulado por la t(11;14) fue recientemente identificado por dos grupos y localizado a 120 kb desde el punto de quiebre MTC. Este gen se denominó PRAD1, porque originalmente se reconoció en el adenoma paratiroideo, pero ha sido oficialmente denominado CCND1. El gen codifica para ciclina D1 y se sobreexpresa en casi todos los casos de LCM, expresándose poco en otros cánceres hematológicos (7).
La sobreexpresión de este gen a nivel del RNA está presente en la mayoría de los LCM, sin cambios en BCL1. Este hecho llevó a detectar, mediante sondas, un punto de quiebre adicional que involucraría al cromosoma 11q13. Todos los puntos de quiebre conocidos dejarían la región codificante de CCND1 estructuralmente intacta, lo que podría aumentar la expresión de proteína. En algunos casos, la pérdida de la secuencia reguladora del extremo 3’ podría aumentar la vida media de la ciclina D1.
Actualmente, la inmunohistoquímica e hibridización in situ para ciclina D1 se pueden hacer en tejidos fijados en parafina.
La sobreexpresión de ciclina D1 produce el acortamiento de la fase G1, probablemente por una interacción física con la proteína del retinoblastoma. La ciclina D1 se une y activa a importantes enzimas, llamadas kinasas, dependientes de ciclina (principalmente CDK4 y CDK6), cuya actividad es importante para avanzar desde G1. El complejo ciclina D1-CDK4 se une a RB fosforilada, que no se une a factores de transcripción, y la célula es promovida a la fase S.
Se ha comunicado la presencia de mutaciones del gen supresor de tumores p53 en variantes agresivas de LCM. Este gen regula la expresión de proteína p21, que es un inhibidor universal del complejo ciclina-CDK, por lo que se sobreexpresa ciclina D1. Si el DNA se daña, p21 se acumula y se inhibe la replicación, para permitir la reparación del DNA. Si no se repara, p53 gatilla la apoptosis, pero si las células tumorales tienen inactivado este gen, no pueden hacer la apoptosis y son genéticamente inestables. Las células acumulan rápidamente mutaciones y alteraciones cromosómicas, lo que conduce a la selección de clones malignos.
Las translocaciones que involucran a los genes 3,8,10,13 y 17 se encuentran con frecuencia en adición a (11;14)(q13q32). Más del 50% de los casos con (11;14)(q13q32) tienen anormalidades cariotípicas adicionales, incluyendo del(11q), del(13q), alteraciones del 3q+12 y deleciones de 6q,1p,9p y 17 p(7). Estudios de hibridización genómica comparativa identificaron regiones de pérdida, ganancia y amplificación en múltiples sitios a nivel cromosomal (7). Entre éstos, la amplificación de la región 3q26.1-29 aparece como predominante (8).
Tratamiento y sobrevida
Antes de reconocerse el LCM como una entidad aparte, los pacientes se agrupaban en linfomas de bajo grado o grado intermedio, de acuerdo a la Working Formulation y recibían varias terapias. Para evaluar éstas, se revisó material histológico de 562 pacientes (9) incluidos en dos estudios de EORTC, identificándose 64 pacientes con LCM (11,4% de los LNH), de los cuales, 29 eran de grado intermedio a alto y 35 eran de bajo grado. Estos grupos fueron comparados en cuanto a sobrevida y respuesta a tratamiento.
Las características clínicas de este grupo confirman que este subtipo de LNH afecta principalmente a gente mayor, que a menudo presentan enfermedad avanzada, con compromiso de médula ósea y hepatoesplenomegalia. Los pacientes con diagnóstico de linfoma de grado intermedio a alto recibieron esquemas con ciclofosfamida, doxorrubicina, teniposido, prednisona, vincristina y bleomicina (CHVmP-VB), o doxorrubicina, ciclofosfamida, etopósido, mecloretamina, vincristina, procarbazina y prednisona (ProMACE-MOPP). El grupo de bajo grado recibió ciclofofosfamida, vincristina y prednisona (CVP) como inducción e IFN como mantención.
Usando el esquema CHOP, la sobrevida estimada a 10 años fue significativamente menor (8%) comparado con otros subtipos histológicos (35%) (1). En general, la sobrevida global fue peor que para otros linfomas de bajo grado, con remisión completa de corta duración. Los pacientes con LCM de grado intermedio a alto que recibieron CHVmP-VB mostraron una sobrevida más larga (65 meses) que los pacientes que recibieron ProMACE-MOPP (33 meses), pero esta diferencia no fue estadísticamente significativa.
La duración de la respuesta tampoco fue muy diferente, pero en la sobrevida sin progresión de enfermedad huvo una leve diferencia. ProMACE-MOPP indujo mayor toxicidad (hematológica y no hematológica). Los pacientes con LCM que recibieron CVP tuvieron sobrevida promedio de 45 meses, mientras que los otros LNH de bajo grado tuvieron sobrevida mayor a 7 años.
La sobrevida promedio es de 3 a 4 años, en algunas series (2), o sea, significativamente menor que en otros pacientes con linfoma. Con el uso de esquemas combinados de quimioterapia sin doxorrubicina, se obtuvo remisión completa en 20 a 40% de los casos (10). La tasa subió a 58% con el uso combinado de ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP), pero las remisiones duraron poco y el porcentaje de curación fue bajo.
Se usaron esquemas de intensificación con bleomicina posterior a la remisión completa o COP (ciclofosfamida, vincristina, prednisona) para los pacientes mayores, con el objetivo de evitar la cardiotoxicidad por antraciclina, con buenos resultados (9). La experiencia con fludarabina e IFN no ha sido alentadora (9).
Como el pronóstico a largo plazo es más bien pobre, también se ha intentado el uso de quimioterapia agresiva más transplante de células progenitoras. Stewart y cols (11) trataron a 9 pacientes con quimioterapia en altas dosis más transplante y encontraron que 3 de ellos no tuvieron progresión de la enfermedad en un plazo de 25 meses.
Majlis y cols (4) analizaron la respuesta al tratamiento y la sobrevida de pacientes no tratados previamente, según su patrón histológico. En el patrón de zona del manto se observó una respuesta favorable a la quimioterapia, con remisión completa en el 75% de los pacientes, con 100% de sobrevida a 3 años y ausencia de progresión de la enfermedad en el 83% de ellos. En cambio, el LCM con patrón de crecimiento difuso se asoció con peor pronóstico, con sólo 20% de remisión completa, 55% de sobrevida a 3 años y ausencia de progresión de la enfermedad en 26%.
La morfología blástica se asoció con peor sobrevida, pero sólo se presenta con patrón de crecimiento difuso. Sólo seis pacientes se clasificaron como variedad nodular, considerándose una etapa intermedia de progresión de la enfermedad, más cercana al patrón difuso, en cuanto a pronóstico y curso clínico.
Factores pronósticos
Existen varios indicadores clínicos y patológicos que sirven como predictores de sobrevida:
- Los predictores clínicos de mal pronóstico son similares a otros linfomas: edad >65 años, performance status pobre (ECOG>2), etapa avanzada, síntomas B, esplenomegalia, compromiso de sangre periférica (linfocitosis), LDH o B2 microglobulina elevadas, albúmina baja, enfermedad residual, anemia, mala respuesta a tratamiento (10).
- Morfológicos: pattern nodular o difuso (contrapartida pattern zona del manto), variante blástica
- Inmunohistoquímicos: Alto índice mitótico (mayor de 2,5 mitosis por campo de aumento mayor).
- Moleculares: Mutación y sobreexpresión del gen p53 e inactivación de p16 (10,12).
Algunos estudios
Onciu y cols. (5) describen los resultados del estudio citogenético de 49 casos bien caracterizados de LCM. De ellos, 30 tenían compromiso leucémico en el momento del diagnóstico y 19 tenían enfermedad nodal y linfocitosis periférica mínima. Se efectuó cariotipo convencional por bandeado G. En el grupo nodal se estudiaron 13 linfonodos y 6 aspirados de médula ósea; en el grupo leucémico se efectuaron 27 aspirados de médula ósea, 2 de linfonodos y 1 estudio de sangre periférica. Se analizaron al menos 20 metafases en 30 casos, 15 a 19 metafases en 11 casos y 3 a 15 en 8 casos.
La edad promedio de los pacientes fue de 65 años (con rango 31 a 85). Hubo 33 hombres y 16 mujeres. De los pacientes con enfermedad nodal, uno tenía linfocitosis moderada, con 4800/ul; en los pacientes con variedad leucémica, el promedio fue 66.000/ul (rango de 11.100 a 647.600). Tres de estos pacientes no tenían diagnóstico definitivo; los restantes tenían los diagnósticos de leucemia linfocítica crónica (14), leucemia linfocítica crónica/leucemia prolinfocítica (2), leucemia prolinfocítica (1), linfoma esplénico con células velludas (1), LCM (7), leucemia linfoblástica aguda (1) y linfoma de células pequeñas no hendidas (1).
El 50% de los pacientes con presentación leucémica y el 37% de los con presentación nodal, recibieron quimioterapia antes del estudio. Sólo el 6% presentaba la t (11;14) como única anormalidad; la anormalidad del cromosoma 13 estaba presente en el 56%, la del cromosoma 17 en 37%, la del cromosoma 8 en el 33% y la del cromosoma 1 en 35% de los casos.
Cohen y cols (3) revisaron los frotis de sangre periférica, aspirado de médula ósea y biopsia de 46 pacientes con LCM, diagnóstico que fue establecido en todos los casos de muestras extramedulares usando morfología estándar, criterios de inmunofenotipo y biología molecular. 27 de 35 pacientes (77%) tenían células de linfoma circulando (con una media de 20% de todos los leucocitos circulantes y rango de 5 a 90%), identificados por morfología en el mismo punto del curso de la enfermedad. No se encontraron diferencias estadísticamente significativas en la sobrevida con o sin compromiso de sangre periférica. El aspirado de médula ósea tuvo un 83% de positividad para linfoma y el de biopsia ósea, 91%.
El patrón de la biopsia ósea fue nodular en 31 casos, intersticial en 19, paratrabecular en 17 y difuso en 12 casos. Aunque la sobrevida promedio de los pacientes con compromiso de MO mayor de 50% fue de 13 meses y con menos de 50% fue 49 meses, no hubo diferencias estadísticas entre estos pequeños subgrupos.
En un estudio de 22 casos realizado en 1990 (5), se encontró que el recuento absoluto de linfocitos mayor de 4.000 era un predictor significativo de sobrevida. Los pacientes que tenían linfocitosis en el momento del diagnóstico tenían una sobrevida de sólo 10 meses, mientras que los que no la tenían tuvieron una sobrevida de 85 meses. Por otra parte, los pacientes con un recuento de plaquetas mayor de 100.000 tenían una sobrevida de 84 meses.
Los hombres tuvieron una sobrevida promedio de 30 meses; las mujeres, de 117 meses. Los pacientes que lograron remisión completa tuvieron sobrevida de 118 meses, cifra que llegó a sólo 75 meses para los que no lo lograron.
Empleando la técnica de Northern blot y la reacción en cadena de polimerasa, Oka y cols (14) investigaron la sobreexpresión del gen PRAD1 en el tejido tumoral de 58 pacientes con linfomas B. La sobreexpresión se detectó en 6 de 8 pacientes con LCM y sólo en 1de 50 casos de otros tipos de linfoma, lo que indica una estrecha relación con LCM. Las muestras de LCM, además, tenían un inmunofenotipo CD5+CD10-IgD+.
Los pacientes con LLC tenían CD5+CD10-IgD+, pero sin sobreexpresión de PRAD1; en cambio, la mayoría de los pacientes con linfoma difuso de bajo grado tenían compromiso extranodal con fenotipo CD5-CD10-IgD-, sin sobreexpresión de PRAD1. Aunque la t(11;14)(q13q32) y el reordenamiento de BCL1 están estrechamente relacionados con LCM, sólo se detectaron en algunos pacientes con LCM. En cambio, en la mayoría de ellos se encontró la sobreexpresión de PRAD1, que permite diferenciarlo de otros tipos de linfoma.
Conclusiones
El LCM es una entidad bien definida desde el punto de vista clínico y anatomopatológico, con características inmunológicas y moleculares distinguibles de otros desórdenes linfoproliferativos.
Es relativamente poco frecuente, pero es importante hacer el diagnóstico, porque la respuesta a los tratamientos es bastante insatisfactoria, lo que ensombrece el pronóstico.
Se requieren terapias más agresivas, como la mieloablación seguida de transplante de stem cells, y nuevas estrategias basadas en anticuerpos monoclonales o en la búsqueda de nuevos blancos moleculares, para mejorar el control de la progresión de la enfermedad.
