Publicado el 1 de octubre de 2009 | http://doi.org/10.5867/medwave.2009.10.4217
Mecanismos de quimiorresistencia en terapia de primera línea de cáncer de ovario epitelial
Mechanisms of chemoresistance in first-line therapy of epithelial ovarian cancer
Resumen
Este texto corresponde a un trabajo de revisión preparado por su autora en el desarrollo del Curso y Seminarios de Oncología Básica, realizado por el Centro de Oncología Preventiva y la Escuela de Postgrado de la Facultad de Medicina de la Universidad de Chile, entre abril y agosto de 2008. El Director del Curso es el Dr. José Manuel Ojeda.
Introducción
El cáncer de ovario epitelial (COE) es la primera causa de muerte por cáncer ginecológico en los Estados Unidos y la tercera en Chile (1, 2). La mayoría de estos cánceres derivan de las células epiteliales de la superficie ovárica, aunque también pueden provenir de otros tipos celulares, como células germinales, células del estroma y células mixtas; y en algunas oportunidades los ovarios son el órgano blanco para metástasis de cáncer de mama o cáncer gástrico, entre otros.
Los eventos moleculares que llevan al desarrollo de cáncer de ovario son desconocidos; un pequeño porcentaje de los casos se asocia a mutaciones germinales en genes BRCA1, BRCA2 y otros, pero las evidencias epidemiológicas y experimentales sugieren que la carcinogénesis en el ovario está predominantemente guiada por factores asociados con la reproducción y la ovulación (3, 4).
Alrededor de 75% de las pacientes con COE se diagnostican en etapas avanzadas de la enfermedad, es decir, cuando ya existe diseminación tumoral fuera de la pelvis y/o linfonodos positivos retroperitoneales o inguinales. El tratamiento estándar de estas pacientes es la cirugía citorreductiva máxima seguida de quimioterapia sistémica (5). En 25% de las pacientes en etapas tempranas la cirugía es el manejo inicial recomendado y sólo en algunos casos se requiere el uso de quimioterapia (6). Los factores pronósticos más importantes en la sobrevida de estas pacientes son la edad en el momento del diagnóstico, el estadio de la enfermedad, el subtipo y grado histológico y el grado de cirugía citorreductiva realizada (16).
Quimioterapia en COE
La quimioterapia de primera línea que se utiliza desde los años 90 en el COE se basa en el uso de platinos, que son los agentes más efectivos en el tratamiento de este tipo de tumor (7), sin diferencias entre cisplatino y carbaplatino. Por otro lado, la administración de taxanos paclitaxel y docetaxel, asociados o en combinación con platino, se asocia a una mejoría significativa en cuanto a respuesta global, remisión clínica completa y mediana de supervivencia libre de enfermedad y global, en comparación con platinos solos (8, 9). Para mujeres con COE avanzado las tasas de respuesta son aproximadamente 90% y 75% alcanza una respuesta clínica completa, es decir, sin lesiones aparentes en imágenes (TAC) o marcadores en sangre (Ca-125). Sin embargo, 75% de este grupo de respuesta clínica completa van a presentar una recaída y eventualmente van a morir por la enfermedad (16). Una de las razones de esto es la quimiorresistencia contra las drogas de primera línea generada por las células tumorales.
La quimiorresitencia es una gran limitante para el éxito del tratamiento de muchos cánceres; esto es particularmente cierto en el cáncer de ovario, donde el desarrollo de quimiorresistencia es un evento muy frecuente. El éxito de la quimioterapia depende del desarrollo del conocimiento de las vías moleculares involucradas y sus mecanismos de integración, lo que permitirá determinar señales de vida o muerte celular sobre las cuales se podrá actuar con nuevos métodos para tratar este gran problema clínico.
Los platinos son agentes alquilantes genotóxicos y su mecanismo de acción es a través de la modificación de las bases nitrogenadas del ADN, lo cual lleva a mutaciones impidiéndose la replicación del ADN, la transcripción de ARN y eventualmente gatillando la apoptosis. Las células que son particularmente sensibles a este tipo de agentes genotóxicos son las que se están dividiendo rápidamente. El cisplatino fue el primer quimioterápico utilizado. Su actividad antitumoral se describió por primera vez en los años 60 y se ha utilizado ampliamente en pacientes con cáncer desde el año 1978; se ha tratado de desarrollar varios análogos del cisplatino con el objetivo de mejorar su eficacia, como el carbaplatino, que se introdujo al uso clínico en 1992; sin embargo, el cisplatino sigue teniendo mayor eficacia contra muchos tumores (10).
El paclitaxel, un derivado de la corteza del árbol Tejo del Pacífico (Taxus brevifolia) demostró actividad antitumoral en 1971. Los taxanos son agentes inhibidores del funcionamiento del huso mitótico, por lo cual interfieren con el mecanismo de división celular. Las fibras que componen el huso mitótico están compuestas por microtúbulos, cuyo funcionamiento requiere de una inestabilidad dinámica en cuanto a la polimerización y depolimerización de la tubulina que los compone; los taxanos estabilizan los microtúbulos, impidiendo este dinamismo. Los microtúbulos se unen y traccionan una de las copias de los cromosomas formados hacia cada lado de la célula en división; sin ello la célula no se puede dividir, la mitosis se detiene en metafase y se provoca la muerte celular.
Algunos mecanismos de quimiorresistencia
Una de las principales causas de falla de tratamiento es el desarrollo de resistencia a la droga por parte de las células cancerosas, lo que genera recurrencias y muertes entre los pacientes con cáncer. Los principales mecanismos generales de resistencia son los siguientes (15):
Selección de células cancerosas resistentes a una droga en particular: considerando que la inestabilidad genética y las tasas de mutación son características claves de las células tumorales, los cambios genéticos que adquieren las células en división son de alta tasa. La heterogeneidad genética de las células tumorales explica porqué no todas las células son sensibles a la misma droga dentro de un mismo tumor. Las células sensibles a una droga dada mueren cuando se exponen a ésta, mientras que las células resistentes a la droga sobreviven y se multiplican, lo que permite el crecimiento de un nuevo tumor resistente a la droga original. Por estas y otras razones es que para el tratamiento de los distintos tumores se ocupan quimioterapias combinadas que atacan distintos procesos celulares, evitando la selección de células tumorales.
Aumento de la expresión de la molécula blanco: las drogas que actúan inhibiendo alguna enzima o proteína claves en el crecimiento y desarrollo celular pueden ver afectado su mecanismo de acción si la célula tumoral aumenta en forma importante el número de dicha molécula, ya que la cantidad de la droga está limitada por su dosificación. Este mecanismo de resistencia tumoral puede operar a través de la amplificación de genes blanco que se replican muchas veces, generando muchas copias de esa región particular del genoma.
Falla de las drogas para entrar a la célula blanco y/o ser arrojadas desde el interior de la célula: las células no alcanzan niveles terapéuticos al interior de la célula por muchas razones; una de las más frecuentes es la amplificación de un gen conocido como MDR1 (Multiple Drug Resistance), que transcribe una proteína transmembrana que impide la entrada de ciertas drogas a la célula o bien las saca de la célula una vez que están adentro. Estas capacidades hacen a esta proteína muy efectiva en la reducción intracelular de distintos agentes quimioterapéuticos.
Impedimento de la droga en alcanzar las células blanco: dependiendo del tamaño y localización de los tumores es posible que las drogas utilizadas no sean capaces de alcanzar su objetivo, fallando así en su acción. Esto ocurre en tumores muy grandes, en los que es difícil alcanzar el centro de la masa por falta de irrigación y en los tumores cerebrales, donde las drogas tienen dificultades para alcanzar los tejidos debido al tipo de circulación y barreras existentes.
Inhibición de la expresión de la molécula blanco: es posible que la molécula blanco desaparezca durante la progresión del desarrollo del cáncer, haciendo que una droga que inicialmente presentaba algún efecto ya no lo tenga.
Alteración de la molécula blanco: debido a que las mutaciones son frecuentes en las células tumorales, un pequeño cambio o alteración en el gen blanco de una droga puede hacer que ésta ya no pueda inhibir su función.
Resistencia a platinos y taxanos en cáncer de ovario epitelial
A pesar de que 80% de los tumores tiene buen respuesta inicial a cisplatino (10, 11), la mayoría de los pacientes recurren con enfermedad resistente. Debido a que platinos y taxanos tienen distintos mecanismos de acción, habitualmente se utilizan combinados en terapias antitumorales. Un artículo de revisión recientemente publicado sugiere que existe una relación inversa entre la resistencia a platinos y taxanos y que la resistencia a una droga hipersensibiliza frente a la otra, de modo que sería beneficioso alternar el uso de estas drogas en el tratamiento clínico del cáncer (10).
Los mecanismos de resistencia encontrados en modelos celulares resistentes a cisplatino incluyen: disminución de la acumulación celular no mediada por glicoproteína P; aumento de los niveles de glutatión transferasa; aumento de la reparación de ADN y aumento de la actividad antiapoptótica. Los mecanismos involucrados en la resistencia a paclitaxel incluyen: disminución de la acumulación celular a través de la glicoproteína P; y alteración en la expresión o modificaciones post-traduccionales de la beta-tubulina o de cualquier proteína reguladora de los microtúbulos.
A pesar del interés que existe en la búsqueda de moléculas para inhibir o evitar la quimiorresistencia relacionada con la no persistencia de las drogas en el interior de la célula o su degradación, muchas de las nuevas terapias en estudio actualmente se concentran en moléculas blanco relacionadas con las vías de la apoptosis y de la proliferación celular. De todos los mecanismos de resistencia expuestos anteriormente, en éstos se ha invertido más esfuerzo y estudio (11, 12, 13, 14).
Rol de los reguladores de la apoptosis en la quimiorresistencia del cáncer de ovario
Rol de Xiap en la modulación de receptores de muerte: Dentro de las proteínas inhibidoras de la apoptosis (IAP) se encuentran las Xiap (proteínas inhibitorias de la apoptosis ligada a X). Los receptores de muerte (DR) son proteínas de superficie de membrana que pertenecen a la superfamilia del factor de necrosis tumoral (TNF). Estos receptores juegan un rol importante en la activación de la vía de la apoptosis a través de la activación de la caspasa 8. Los más estudiados son Fas, TNF Receptor 1 y 2 (TNR1, TNFR2) y el receptor TNF relacionado con el ligando inductor de apoptosis (TRIAL). Xiap tiene la función de inhibir directamente la caspasa 9 (iniciador de la apoptosis a través de la vía intrínseca) y a las caspasas 3 y 7 (efectoras de la activación de ambas vías de la apoptosis). De esta manera Xiap está implicada en la promoción de la supervivencia celular por medio de la modulación de receptores de muerte e inhibiendo directamente a las caspasas efectoras 3 y 7. Se ha comunicado que el cisplatino produce una disminución de Xiap en células quimiosensibles de cáncer de ovario, lo que no se observa cuando existe quimiorresistencia.
Rol de FLIP en la modulación de receptores de muerte: TNFalfa, FasL y TRIAL son miembros de una gran familia de citoquinas citotóxicas de ciertas células tumorales. La proteína inhibitoria tipo-FLICE (FLIP, FLICE se le dice a la caspasa 8 inactiva) puede interferir con la activación de la apoptosis inhibiendo la activación de la caspasa 8. Se ha sugerido que la resistencia de las células de cáncer de ovario está relacionada con la inducción de FLIP para disminuir el efecto citotóxico de TNFalfa y TRIAL. De esta manera FLIP interfiere con las vías de muerte mediada por citoquinas de estas células tumorales, confiriendo resistencia a los agentes quimioterapéuticos.
Xiap y FAK en quimiorresistencia: Focal-Adhesion Kinase (FAK) es una proteína tirosina kinasa no asociada a receptor que se activa al asociarse con integrina, suprimiendo la apoptosis a través de la activación de otras kinasas. El cisplatino provoca la rotura de esta molécula inactivándola. La sobreexpresión de Xiap previene esta acción del cisplatino haciendo a la célula resistente y alterando además la adhesión celular.
P53 en quimiorresistencia mediada por Xiap y Akt: Debido a la función que cumple la proteína p53 en la reparación del ADN y en la apoptosis, se ha sugerido que defectos en esta proteína podrían determinar resistencia a la quimioterapia. La mutación del gen p53 esta presente en la mayoría de los cánceres, detectándose aproximadamente en la mitad de los cánceres de ovario avanzado (13). Cuando se produce daño celular se activa una serie de kinasas que fosforilan a la p53, aumentando su acción transcriptacional. El rol que esta proteína cumpla va a depender del tiempo de exposición al daño que haya soportado la célula; frente a un daño celular leve p53 expresará proteínas relacionadas con reparación y si el daño es grave se sobreexpresarán genes regulados por p53, relacionados con la inducción de apoptosis.
Las AKT son proteína kinasas activadas por otras kinasas (PI3K). La activación de AKT es el único blanco de PI3K asociado con transformaciones malignas. Altos niveles de AKT/PI3K han sido vinculados con mal pronóstico y quimiorresistencia en cáncer de ovario, tanto para cisplatino como, en un reporte reciente, para paclitaxel. Se ha sugerido que existe un feedback en la regulación de Xiap y AKT: mientras Xiap aumenta la fosforilación de AKT y requiere de AKT para su función, Xiap es protegida del down-regulation por cisplatino. Varios estudios han establecido que a través de la fosforilación de Murine Double Minute (MDM2), proteína encargada de limitar la actividad de regulación transcriptacional de la p53, por AKT, se mantienen niveles bajos de p53, aunque no se sabe por qué mecanismo exacto el cisplatino no logra aumentar la up-regulation de esta proteína en células quimiorresistentes (12).
En la siguiente imagen se ilustra lo expuesto. En una célula quimiosensible de cáncer de ovario (A) el cisplatino aumenta el contenido de p53, llevando a la célula a una mayor síntesis de proteínas que promueven la detención del ciclo celular, como la p21, y proteínas proapoptóticas como Bax y Fas. Estas últimas activan tanto la vía intrínseca (mitocondrial) como la extrínseca (receptor de muerte) apoptóticas, cuyo resultado final es la activación de la activación de las caspasas-3 (y -7, dato no mostrado). En estas células, los mediadores de supervivencia celular como Xiap, AKT y Flip (en rojo en la figura) están menos expresados o están en un estado inactivo. La prohibitina puede también jugar un rol en la inhibición de la progresión del ciclo celular a través de la vía de la proteína Rb-E2F, uniéndose a la Rb. Al contrario, en una célula quimiorresistente (B) el aumento de la ubiquitinización de p53 por MDM2 resulta en una mantención de bajos niveles de p53, a pesar de la presencia de cisplatino, el cual además no es capaz de generar la disminución de Xiap, y por lo tanto de la mantención de la actividad de la vía PI3K/Akt. Sumándose a esto, la unión de TNFR2 por TNFalfa lleva a un aumento de los niveles de FLIP a través de la vía de NFkappaB, inhibiendo así la acción proapoptótica de las citoquinas a través de TNFR1. En suma, como una consecuencia de la falla en la activación la cascada de las caspasas en respuesta a un agente quimioterapeútico, estas células pierden su capacidad de entrar en apoptosis, y es así como llegan a ser células quimiorresistentes (Fig. 1).
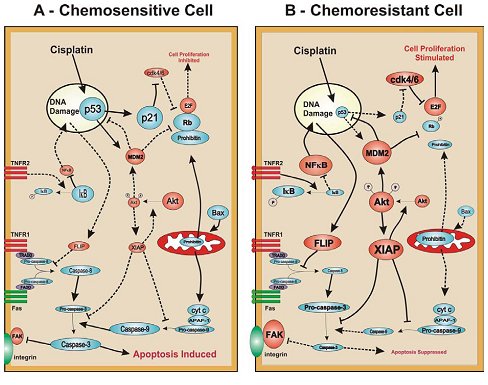 Tamaño completo
Tamaño completo Conclusión
Existen muchas vías y mecanismos involucrados en la generación de quimiorresistencia en terapia de primera línea de cáncer de ovario epitelial. Los avances en el conocimiento y entendimiento de éstos permitirán que en el futuro se desarrollen quimioterapias exitosas.
