Acta de reunión clínica
← vista completaPublicado el 1 de febrero de 2009 | http://doi.org/10.5867/medwave.2009.02.3797
Enfrentamiento clínico de la talla baja
Clinical management of short stature
Resumen
Este texto completo es la transcripción editada de una conferencia dictada en el marco de las reuniones clínicas de la Unidad General de Cuidados del Niño del Hospital Padre Hurtado. La publicación de estas actas científicas ha sido posible gracias a una colaboración editorial entre Medwave y la Unidad. El jefe de la UGCN es el Dr. Alejandro Donoso y el Encargado de las Reuniones Clínicas es el Dr. Mario Vildoso.
Definición de talla baja
Para establecer que un paciente tiene talla baja es muy importante medirlo en forma correcta y usando el instrumento adecuado: infantómetro para menores de 3 años o niños mayores que no hayan logrado la bipedestación y estadiómetro para niños de más de 3 años; y luego analizar este resultado según sexo y edad, en tablas adecuadas a la población a la que pertenece el niño.
Hay varias situaciones en las que se plantea el problema de la talla baja:
- Talla absoluta bajo dos desviaciones estándar (-2DS) o bajo el percentil 3 (p3) de las tablas norteamericanas, lo cual no es lo óptimo, ya la constitución genética y las condiciones de vida son diferentes. Actualmente en Chile se usan las tablas de la OMS para niños de 0 a 6 años y luego las tablas CDC.
- Discordancia con talla target: se habla de talla baja cuando la talla del paciente es al menos 1 DS más baja que la talla target (talla diana), aunque no quede en rango de talla baja según el criterio anterior.
- Baja velocidad de crecimiento, o cuando el paciente cambia de carril en la curva de crecimiento.
- Expectativa social: se presenta frente a condiciones en que un paciente crece sobre percentil 3, adecuado a su carga genética y con buena velocidad de crecimiento, sin embargo tiene expectativas sociales más exigentes, como pertenecer a un equipo de básquetbol, etc.
En un paciente con talla baja se debe evaluar otros parámetros en el examen físico, además de la talla propiamente tal. A continuación se describe con detalle estos parámetros.
- Perímetro cefálico: está aumentado en ciertas patologías genéticas, como la acondroplasia.
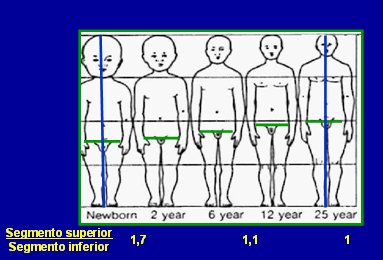
- Relación entre los segmentos superior e inferior: se mide la talla total y la distancia pubis-suelo; de la diferencia entre ambas se obtiene la medida del segmento superior; luego se obtiene la relación SS/ SI. Si el paciente tiene talla baja con desproporción de segmentos es probable que se trate de un síndrome genético (diaplasias esqueléticas). Al final de esta lista se ilustra este punto.
- Envergadura: debe ser igual a la talla o tener una variación menor de 5% respecto a ésta.
- Talla diana (target): permite verificar que el paciente esté creciendo de acuerdo con su potencial genético. Si se trata de un niño, debería medir el promedio de la talla de ambos padres, nivelando a la mujer como si hubiese sido hombre con la suma de 13 cm. En caso de ser niña, la talla diana se obtiene de la talla de la madre más la talla del padre menos 13 cm, dividido por dos.
- Edad ósea: permite diferenciar dos condiciones fisiológicas que cursan con talla baja, la talla baja constitucional y la talla baja familiar: en la talla baja constitucional la edad ósea está retrasada y en la familiar, es adecuada. Patologías como el hipotiroidismo y la deficiencia de hormona del crecimiento (GH) también producen retraso en la edad ósea, de modo que esta condición siempre se debe analizar en relación a la clínica.
- Dentición: se asocia a madurez, por lo tanto se debe preguntar cuándo aparecieron los primeros dientes y cuándo se produjeron los cambios de los dientes temporales.
- Desarrollo puberal: no es lo mismo una talla para una edad determinada en un paciente con desarrollo de Tanner grado 1 que grado 3, ya que lo que le queda por crecer es diferente en ambos casos.
- Peso al nacer: entre 10 y 20 % de los niños pequeños para la edad gestacional (PEG) no hacen un crecimiento compensador, permaneciendo bajo p3. Estos niños se benefician de terapia con hormona de crecimiento, por lo que es aconsejable que se mantengan en control pediátrico y si a los 4 años no se han logrado ubicar en su canal de crecimiento según la carga genética, se deriven a endocrinología para eventual tratamiento con GH.
En la siguiente imagen se ilustra lo descrito en el punto 2: el cambio en la proporción de los segmentos corporales que ocurre durante la vida del ser humano, desde recién nacido hasta la edad adulta.
 Tamaño completo
Tamaño completo El mecanismo que explica el crecimiento tiene su punto de partida en el cerebro, con la liberación de al menos dos péptidos hipotalámicos: uno estimulador, GHRH y uno inhibidor; somatostatina. La interacción entre éstos determina la pulsatilidad de la secreción de la Hormona de Crecimiento (GH). Esta hormona actúa a nivel óseo y de los demás tejidos a través del sistema IGF-I (Insulin-Like Growth Factor I), produciendo el crecimiento. Una alteración en cualquiera de estos niveles puede producir fallas en el proceso de crecimiento del individuo; por ello, frente a un paciente que consulta por talla baja se debe tener claro las posibles etiologías y proceder a descartarlas en forma ordenada.
Etiología de la talla baja I: variantes normales
Las variantes normales del crecimiento que se pueden presentar como talla baja son:
- Talla baja familiar
- Retraso de crecimiento constitucional
Talla baja familiar: En este caso el paciente tiene antecedentes familiares de talla baja; no tiene alteraciones sistémicas ni endocrinas; tiene peso y talla normales al nacer y durante los primeros dos años de vida se va situando en su carril de crecimiento, para luego caer bajo lo normal; su edad ósea es concordante con su edad cronológica; y el desarrollo de sus características sexuales es concordante con su edad, es decir, tiene una pubertad normal.
Retraso constitucional: En el retraso de crecimiento constitucional, en 60 a 90% de los casos hay antecedente familiar de menarquia tardía en la madre o “estirón” tardío en el padre; el peso y talla son normales al nacer, no hay historia de enfermedad sistémica ni endocrina, el estado nutritivo es normal y en el examen físico no hay alteración de los segmentos corporales. Estos niños tienen edad ósea retrasada con respecto a la cronológica y retraso puberal. Por lo general alcanzan la talla diana, pero no siempre (Fig.2).
 Tamaño completo
Tamaño completo II. Alteraciones endocrinas
Las alteraciones endocrinas que pueden cursar con talla baja son:
- Hipotiroidismo, congénito o adquirido.
- Alteraciones de eje somatotrófico, sea deficiencia de hormona del crecimiento (GH) o resistencia a ésta.
- Exceso de glucocorticoides.
- Otras alteraciones endocrinas.
Hipotiroidismo congénito: se detecta mediante screening neonatal, de modo que ante un paciente con hipotiroidismo congénito con talla baja se debe buscar otra causa o pensar en negligencia materna en cuanto a la administración de la hormona. El hipotiroidismo adquirido se puede presentar a cualquier edad; el hecho de que una persona no haya presentado hipotiroidismo congénito en el screening no significa que no pueda desarrollar hipotiroidismo adquirido en el transcurso de su vida, por lo general en la edad escolar o adolescente.
Deficiencia de GH: puede ser congénita o adquirida.
- La deficiencia de GH congénita puede ser: aislada; acompañada de deficiencia de otras hormonas hipofisiarias, por lo tanto frente a un niño con déficit de GH es indispensable evaluar todos los ejes de esta glándula, incluyendo la neurohipófisis; por defectos de la línea media, como hipoplasia del tercio medio o fisura labiopalatina; por agenesia hipofisiaria; y asociada a deficiencia de genes.
- La deficiencia adquirida puede ser secundaria a: tumores hipotálamo-hipofisiarios; histiocitosis de Langerhans; infecciones o granulomas del SNC; trauma de cráneo que comprometa la hipófisis; hidrocefalia; silla turca vacía; e hipofisitis autoinmune, una inflamación de la glándula que lleva a deterioro de la función. Finalmente, la deprivación psicosocial puede producir deficiencia de GH sin un sustrato orgánico que la explique; en estos casos, cuando se retira al niño del medio adverso empieza a crecer de manera normal.
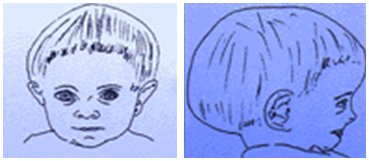
Resistencia a GH: se ve en el síndrome de Laron, que se caracteriza por resistencia a la GH. En los exámenes se encuentra GH alta con IGF-1 baja. En la Fig. 3 se muestra un dibujo que representa a un niño con deficiencia de GH: tiene hipoplasia del tercio medio facial y frente amplia. En estos niños el peso y la talla son normales al nacer, pero la velocidad de crecimiento disminuye en general a partir de los seis meses de edad; los varones pueden tener micropene. Además en el período neonatal pueden presentar hipoglicemia, debido a que la GH es una hormona de contrarregulación, e ictericia colestásica neonatal.
 Tamaño completo
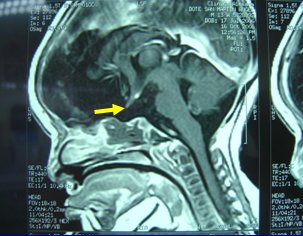
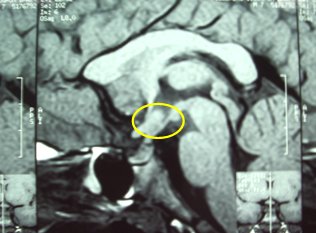
Tamaño completo La siguiente resonancia magnética (RM) corresponde a un niño que presentó hipoglicemia neonatal grave e ictericia colestásica asociadas a defecto de la línea media, fisura labiopalatina y agenesia de la hipófisis. La mancha hiperintensa corresponde a la neurohipófisis, que siempre se debe evaluar (Fig. 4).
 Tamaño completo
Tamaño completo A continuación se muestra una RM de otro paciente, portador de un tumor hipotalámico que destruyó el tallo hipofisiario, produciendo un déficit completo de las hormonas de la adeno y la neurohipófisis. Este niño consultó a los 7 años de edad por signos de pubertad precoz, pero no tenía el aspecto de un niño de 12 a 14 años, sino que era pequeño, delgado y con desarrollo sexual grado 4 de Tanner, lo cual era absolutamente discordante. La evaluación demostró que se trataba de un tumor que provocaba todas las deficiencias: diabetes insípida, déficit de ACTH e hipotiroidismo central. El diagnóstico final fue germinoma del hipotálamo. La similitud entre la beta-hCG y la LH hizo que este niño produjera testosterona a nivel testicular sin tener activo el eje hipotálamo-gonadal (Fig. 5).
 Tamaño completo
Tamaño completo Exceso de glucocorticoides: también produce talla baja. El hipercortisolismo puede ser endógeno o exógeno, siendo este último el más frecuente, bajo la forma de un síndrome de Cushing secundario al uso de corticoides como tratamiento de patologías inflamatorias crónicas.
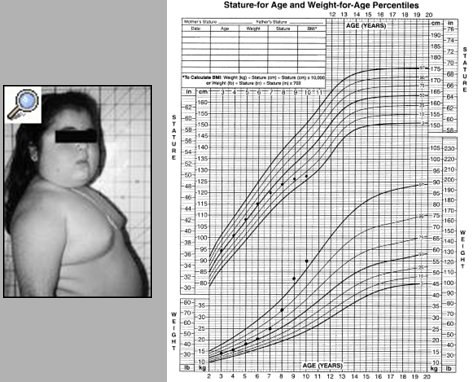
Otras alteraciones endocrinas: la diabetes mellitus mal controlada, el raquitismo hipofosfémico y la hiperplasia suprarrenal congénita, que se caracteriza por déficit de 21 y 11 hidroxilasa y exceso de andrógenos, con aumento de la edad ósea. Estos niños son altos durante la infancia, pero tienen talla baja en la edad adulta debido a que los cartílagos de crecimiento se cierran prematuramente por una pubertad precoz. En la siguiente imagen se observa una niña con síndrome de Cushing, que se caracteriza por disminución de la velocidad de crecimiento, aumento de peso, edad ósea atrasada, hipertensión arterial, cara de luna, dorso de búfalo y estrías violáceas. La principal causa del exceso de corticoides es la iatrogenia; causas raras son el adenoma productor de ACTH y el tumor de glándula suprarrenal productor de cortisol (Fig. 6).
 Tamaño completo
Tamaño completo III. Displasias esqueléticas y enfermedades de depósito
En este grupo de patologías se encuentran:
- Acondroplasia
- Osteogénesis imperfecta
- Enfermedades de depósito
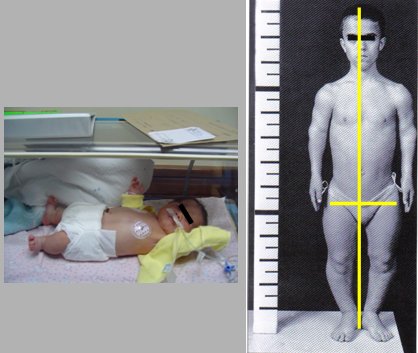
Acondroplasia: se caracteriza por peso normal y talla baja al nacer; la talla baja es desproporcionada, tienen frente prominente, puente nasal bajo y cabeza grande en relación al cuerpo; la talla adulta es 132 cm en hombres y 124 cm en mujeres. En la siguiente imagen se observa un niño de tres días de vida en el que se aprecia claramente el acortamiento del segmento inferior. Estos niños no tienen déficit hormonal, pero se han tratado con GH con resultados discordantes; algunos autores refieren que es útil y otros afirman que no sólo no sirve, sino que además acentúa la desproporción corporal (Fig. 7).
 Tamaño completo
Tamaño completo Enfermedades de depósito: las mucopolisacaridosis se caracterizan por facies tosca, macrocefalia y deterioro de la curva de crecimiento, por lo general a partir del segundo año de vida. Además se puede encontrar valvulopatía, miocardiopatía, hepatoesplenomegalia, mano en garra por contractura articular, compromiso esquelético, que se conoce como disostosis múltiple, retraso global del desarrollo sicomotor y pérdida progresiva de las habilidades motoras e intelectuales adquiridas.
IV. Síndromes genéticos asociados a talla baja
Existen múltiples síndromes genéticos que se asocian a talla baja. Los principales son:
- Turner
- Silver Russel
- Noonan
- Otros
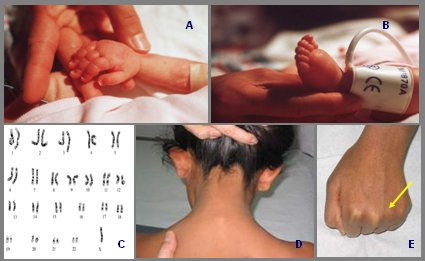
Síndrome de Turner (45,X): Se debe tener siempre en mente porque se puede presentar de diversas maneras. Se puede diagnosticar al nacer, pero muchas veces se diagnostica en la adultez. En el recién nacido se puede manifestar con linfedema de extremidades y cardiopatía congénita; en niñas mayores con diagnóstico de talla baja se debe buscar cúbito valgo, implantación baja del cabello en forma de tridente y acortamiento del cuarto metacarpiano que se hace evidente al empuñar la mano, así como paladar ojival, mamilas separadas, tórax ancho y disgenesia gonadal, todos signos que orientan a este síndrome (Fig. 8).
 Tamaño completo
Tamaño completo Síndrome de Silver Russel: se caracteriza por bajo peso para la edad gestacional, pseudohidrocefalia, frente prominente, cierre tardío de la fontanela anterior, cara pequeña y triangular, asimetría total o parcial y clino o clinobraquidactilia de quinto dedo.
Síndrome de Noonan: es similar al síndrome de Turner, pero en varones: son niños con cara triangular, hipertelorismo, cuello corto, implantación baja del cabello, talla baja, cardiopatía congénita y, en 50% de los casos, estenosis pulmonar. Además pueden presentar cúbito valgo, visión anormal, hepatoesplenomegalia, testes no descendidos, retraso mental leve y microcefalia (Fig. 9).
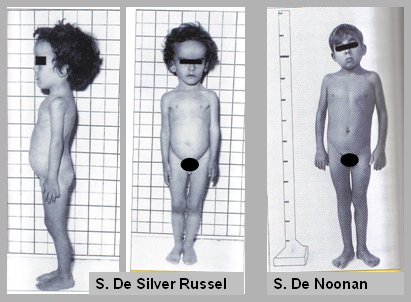 Tamaño completo
Tamaño completo Otros síndromes genéticos con talla baja: trisomías 13, 18 y 21 y síndrome de Prader Willi.
V. Enfermedades crónicas y malnutrición
Las enfermedades crónicas de corazón, pulmón, riñones, gastrointestinales, hepáticas, hematológicas e infecciones crónicas también se asocian a talla baja. Las enfermedades gastrointestinales, específicamente en la enfermedad celíaca, se pueden presentar con talla baja como único signo, sin alteración de las deposiciones.
La malnutrición por la causa que sea puede causar talla baja, (anorexia nerviosa, desnutrición, deficiencia de hierro, deficiencia de zinc, anorexia por quimioterapia para neoplasias, tratamiento con anfetaminas, etc). Con respecto a la deficiencia de zinc, la evidencia ha mostrado que los niños que están bien nutridos no tienen deficiencia de este elemento.
Estudio del paciente con talla baja
Frente a un paciente con talla baja, el aumento del índice peso/talla y del IMC orienta a patología endocrina, aunque también se puede encontrar esto en un paciente con talla baja por variante normal del crecimiento y obesidad exógena (que es cada día más común en nuestro medio). Un índice peso/talla e IMC bajo orienta a patología sistémica y en presencia de dismorfias se debe pensar en un síndrome genético (Fig. 10).
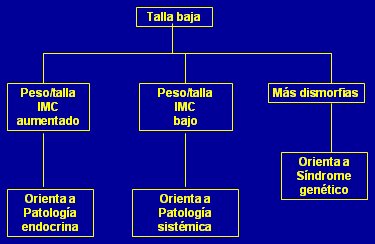 Tamaño completo
Tamaño completo Se debe solicitar los siguientes exámenes para descartar las patologías sistémicas que se mencionan. Estos exámenes deberían estar en rangos normales para apoyar el diagnóstico de talla baja constitucional o familiar:
- Hemograma y VHS: anemias crónicas, poco frecuentes en nuestro medio, enfermedades inflamatorias intestinales.
- Perfil bioquímico: insuficiencia hepática, renal, alteraciones del metabolismo calcio-fósforo.
- Orina completa y urocultivo: la infección urinaria puede ser causa de mal incremento pondoestatural en lactantes.
- Gases en sangre venosa y electrolitos plasmáticos: acidosis tubulares renales.
- Examen parasitológico de deposiciones: giardiasis u otras parasitosis que causan malabsorción.
- Radiografía de carpo, edad ósea (EO): retraso constitucional. También se puede encontrar EO menor a la cronológica en enfermedades endocrinas como hipotiroidismo y deficiencia de GH.
- TSH y T4 libre: para descartar hipotiroidismo se debe evaluar TSH y T4L, ya que si se solicita solamente TSH ésta puede estar en rango normal en hipotiroidismo central. El hipotiroidismo primario, por alteración tiroidea se caracteriza por TSH alta con T4L baja. El hipotiroidismo central, por alteración hipotálamo-hipofisiaria, da THS normal o baja con T4L baja.
El estudio específico, orientado según hallazgos, incluye:
- Anticuerpos antitransglutaminasa: enfermedad celiaca.
- Cariotipo: síndromes genéticos.
- IGF-I, IGFBP-3.
- Test de estímulo para GH: se solicita cuando existe sospecha de alteración del eje somatotrófico.
Algoritmo de estudio de talla baja
Si la historia clínica y las mediciones antropométricas muestran un paciente con talla baja (< p3 con velocidad de crecimiento bajo p5), lo primero es determinar el IMC. Si éste es bajo se debe investigar la presencia de enfermedad crónica o malnutrición.
Si la talla baja está sólo un poco por debajo de p3 (o sea entre -2DS y -3DS) y la velocidad de crecimiento es adecuada, lo más probable es que se trate de una variación normal, ya que las patologías se concentrar en tallas bajo -3DS (Fig. 11).
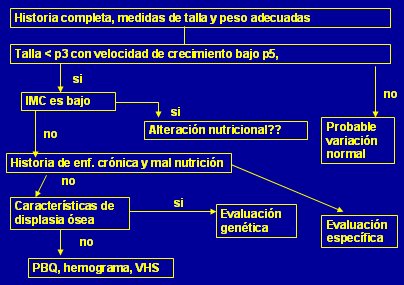 Tamaño completo
Tamaño completo En la práctica clínica los exámenes iniciales incluyen hemograma, VHS, perfil bioquímico, gases venosos, electrolitos plasmáticos, orina completa, urocultivo, examen parasitológico de deposiciones, TSH y T4L. Los resultados se pueden analizar según el siguiente algoritmo (Fig. 12).
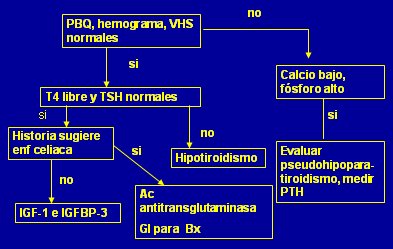 Tamaño completo
Tamaño completo Como segunda línea se puede solicitar anticuerpos antitransglutaminasa y estudio del eje somatotrófico. Si IGF-I e IGFBP-3 no son normales se debe reevaluar el estado nutricional, porque IGF-I disminuye cuando el paciente está desnutrido, se produce un estado de resistencia a GH. En caso de que IGF-I sea baja y la nutrición sea adecuada se procede a efectuar pruebas de estímulo de GH; si no se obtiene un peak adecuado se diagnostica deficiencia de GH y el paciente será candidato a uso de hormona de crecimiento (Fig 13).
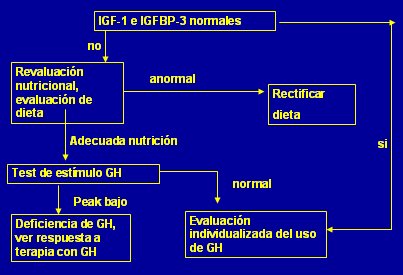 Tamaño completo
Tamaño completo La derivación al especialista se debe efectuar en los siguientes casos:
- Talla menor de 3 desviaciones estándar.
- Talla discordante con la carga genética.
- Disminución de la velocidad de crecimiento.
- Dismorfias y alteración de los segmentos corporales.
