Revisión clínica
← vista completaPublicado el 1 de julio de 2011 | http://doi.org/10.5867/medwave.2011.07.5068
Patología de hígado graso no-alcohólico (HGNA) asociada a obesidad: mecanismos patogénicos
Pathology of nonalcoholic fatty liver disease (NAFLD) associated with obesity: pathogenetic mechanisms
Resumen
La enfermedad de Hígado Graso No-Alcohólico (HGNA) es la causa más importante de enfermedad hepática crónica y es considerada la manifestación hepática del síndrome metabólico asociado a obesidad y diabetes mellitus tipo 2. Se asocia a un amplio espectro de daño hepático, que incluye tanto la esteatosis simple o acumulación intracelular de triacilglicéridos (TAGs), como la inflamación, fibrosis y cirrosis (esteatohepatitis no-alcohólica). Los mecanismos implicados son de carácter multifactorial, siendo la resistencia a la insulina un factor común que genera retención de ácidos grasos y TAGs dentro de los hepatocitos, con la producción de radicales libres a nivel mitocondrial capaces de inducir estrés oxidativo, producción de citoquinas y necrosis. Concomitantemente, se observa baja biodisponibilidad hepática de ácidos grasos poli-insaturados de cadena larga de la serie n-3, lo que alteraría la expresión de factores de transcripción asociados a la lipólisis y lipogénesis a nivel hepático. Un mejor conocimiento de los mecanismos etiopatogénicos del HGNA es fundamental para el desarrollo de estrategias terapéuticas eficaces a futuro.
Introducción
En la actualidad, la enfermedad de Hígado Graso No-Alcohólico (HGNA) es la causa más importante de enfermedad hepática crónica relacionada al aumento en la incidencia de obesidad y diabetes mellitus tipo 2 en la población1. El HGNA se caracteriza por la presencia de esteatosis simple o acumulación intracelular de triacilglicéridos (TAGs), que puede progresar a la inflamación, fibrosis y cirrosis (EsteatoHepatitis No-Alcohólica, EHNA)2. El HGNA es considerado la manifestación hepática del síndrome metabólico, y su prevalencia en la población general alcanza el 15-20%, con una incidencia del 3% de EHNA. En sujetos con diabetes mellitus tipo 2 la incidencia es cercana al 50%, en la población obesa es de 76 a 90%, de los cuales alrededor del 35% desarrollará EHNA, mientras que la EHNA se presenta casi en la totalidad de los obesos mórbidos con diabetes2 3. La biopsia hepática es un factor clave para el diagnóstico del HGNA, que permite distinguir entre esteatosis simple, EHNA y grado de fibrosis. Sin embargo, existen técnicas como el ultrasonido, la tomografía computarizada o la resonancia magnética mediante las cuales se puede confirmar la presencia de esteatosis hepática con un alto grado de precisión2 4. Para su diagnóstico se requiere la exclusión del consumo de 20g/día de alcohol para las mujeres y 30g/día para los hombres, junto con descartar enfermedades virales, respuestas autoinmunes, factores hereditarios o metabólicos, drogas y toxinas5. El desarrollo de la resistencia a la insulina (RI) y del estrés oxidativo se consideran como los principales factores patogénicos del HGNA, los cuales, con la concurrencia de factores nutricionales, pueden determinar el inicio de la esteatosis y su progresión a la EHNA1 2 5. Las implicaciones patogénicas de estos factores son discutidos en este artículo de revisión, con el objeto de identificar mecanismos moleculares claves que pudiesen ser susceptibles de modificar por medio de intervenciones terapéuticas.
Alteraciones metabólicas, estrés oxidativo y resistencia a la insulina asociados a obesidad
Los mecanismos implicados en la acumulación de TAGs a nivel hepático y el subsiguiente daño hepatocelular son de carácter multifactorial y su naturaleza se está empezando a establecer. En condiciones normales, los ácidos grasos (AGs) son el principal combustible para el hígado. Sin embargo, en patologías como la obesidad, la gran afluencia de hidratos de carbono y lípidos induce cambios significativos en el metabolismo intermediario en el hígado.
Los elevados niveles de insulina (hiperinsulinemia) no son capaces de suprimir el flujo de AGs, mostrando un importante nivel de resistencia periférica a la acción de la insulina. El aumento en el pool de AGs circulantes es uno de los principales factores determinantes en la patogénesis del HGNA.
Es así como la hiperglicemia e hiperinsulinemia asociadas promueven la síntesis de AGs a partir de la glucosa e inhiben la β-oxidación de AG, siendo estos AGs re-direccionados hacia la formación de TAGs1 2 5. Considerando que la cantidad de AGs que son exportados vía lipoproteínas de muy baja densidad (VLDL) está limitada por la síntesis de su componentes proteicos, el exceso de AGs es principalmente convertido en TAGs y depositados en los hepatocitos con el consumo de dietas hipercalóricas. Debido a que el hígado tiene una capacidad limitada para la acumulación de TAGs, el depósito de lípidos en condiciones de sobrealimentación determina elevados niveles de AGs saturados, los que se asocian a disfunción y muerte celular. En efecto, estudios experimentales indican que el exceso de AGs condiciona altas tasas de b-oxidación, con producción de especies reactivas del oxígeno (EROS: radical superóxido [O2-] y peróxido de hidrógeno [H2O2]) a nivel de la cadena respiratoria mitocondrial, concomitantemente con la inducción de necrosis6. Estos resultados sugieren que la sobrealimentación puede inducir la sobrecarga de AGs en el hígado provocando altas velocidades de b-oxidación y formación de EROS, lo que está de acuerdo con los cambios en los parámetros relacionados con estrés oxidativo observados en el hígado de pacientes obesos con esteatosis7 10 (Figura 1). En efecto, en forma relativa a los valores controles, el hígado de los pacientes obesos presenta:
- Disminución en el potencial antioxidante (menor actividad de superóxido dismutasa y contenido de glutatión).
- Aumento en la actividad pro-oxidante (mayor lipoperoxidación, contenido de hidroperóxidos y oxidación de proteínas).
- Activación de las células de Kupffer (mayor producción del radical superóxido y tasa lipoperoxidativa), parámetros asociados a la disminución de la capacidad antioxidante del plasma e incremento en los niveles de F2-isoprotanos séricos (productos de la peroxidación del ácido araquidónico) (Figura 1).
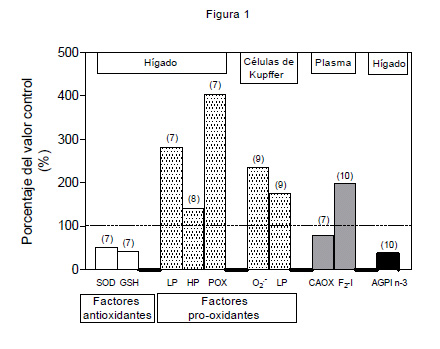 |
| Figura 1: Parámetros asociados al estrés oxidativo en pacientes obesos con hígado graso no-alcohólico, expresados como porcentaje de los valores controles. Los números entre paréntesis corresponden a las referencias específicas que contienen los datos. |
El desbalance redox observado en pacientes obesos representa un fenómeno de estrés oxidativo nutricional, que resulta de la ingesta excesiva y prolongada de combustibles metabólicos (carbohidratos y lípidos) y/o suministro inadecuado de antioxidantes dietarios11.
En condiciones de estrés oxidativo hepático, los pacientes obesos exhiben dos importantes alteraciones asociadas a este desbalance redox:
- Desarrollo de RI12 evidenciada por el aumento del índice HOMA que excede en más del 100% el valor normal (Figura 2)1, y
- Pérdida sustancial del contenido hepático de los ácidos poli-insaturados n-3 (AGPI n-3)10 13, los ácidos eicosapentaenoico (EPA) y docosahexaenoico (DHA) (Figura 1).
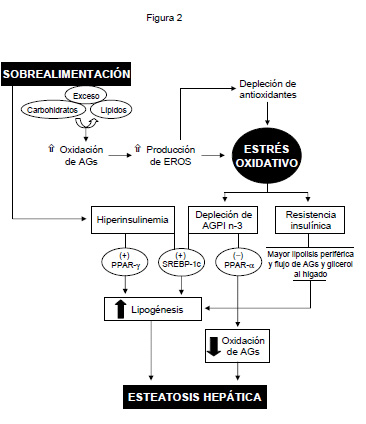 |
| Figura 2: Inducción de estrés oxidativo y su relación con la resistencia a la insulina (RI) y la esteatosis en el hígado graso no-alcohólico asociado a obesidad. |
Pérdida del control de la distribución metabólica de los ácidos grasos asociada a la obesidad
El estrés oxidativo asociado a obesidad tendría un papel causal en el desarrollo de la RI, condición en la cual la captación de glucosa por el músculo y tejido adiposo y la inhibición de la producción de glucosa por el hígado no responden adecuadamente a la insulina, mientras que los efectos pro-lipogénicos de la insulina son mantenidos a nivel hepático14. Este fenómeno se asocia a un cambio en el tipo de fosforilación del receptor de insulina y sus sustratos, que normalmente ocurre en residuos de tirosina, a residuos de serina, mediada por la activación de diversas serina-quinasas activadas por estrés oxidativo15. Esta modificación específica disminuye la fosforilación del receptor de insulina en tirosina y la vía de señalización intracelular de la hormona gatillando la RI12, alteración que podría ser facilitada por la depleción de AGPI n-3, por pérdida del grado de insaturación de los fosfolípidos de membrana que es requerido para el funcionamiento normal del receptor de insulina1.
Los AGPI n-3 ejercen sus efectos regulando el metabolismo de los lípidos en el hígado vía modificación en la transcripción génica, ya sea inhibiendo la expresión y procesamiento del factor pro-lipogénico proteína ligante al elemento regulador de esteroles-1c (SREBP-1c), y activando al receptor activado por proliferadotes peroxisomales-a (PPAR-a) que controla la expresión de los genes de enzimas de la oxidación de los AGs1 16. La disminución del contenido hepático de los AGPI n-3 en la obesidad se ha relacionado con:
- La mayor oxidación de EPA y DHA por el estrés oxidativo inducido (Figura 2)
- La ingesta deficitaria de EPA, DHA y su precursor esencial el ácido a-linolénico13
- La alteración en la síntesis hepática de AGPI n-3 debida a la menor actividad de la D-5 y D-6 desaturasas asociada a la RI17, y
- A la mayor ingesta de AGs insaturados de tipo trans que inhiben drásticamente a la D-6 desaturasa13.
Desde el punto de vista funcional, la depleción de los AGPI n-3 favorecería la esteatosis hepática al desactivar PPAR-a, lo cual disminuye la capacidad de oxidación de AGs, conjuntamente con incrementar la capacidad lipogénica del hígado al activar a SREBP-1c18 (Figura 2). De acuerdo con estos cambios, se ha observado que los pacientes obesos hiperinsulinémicos presentan mayor lipogenesis de novo19 20, proceso que estaría favorecido por la concomitante activación del factor pro-lipogénico PPAR-g21, además de SREBP-1c18, y la mayor lipólisis periférica asociada (Figura 2)1 22. Debido a la desactivación de PPAR-a relacionada con la depleción de los AGPI n-3 hepáticos en la obesidad, se produce un aumento significativo en la relación SREBP-1c/PPAR-a18, lo que determina un desbalance metabólico entre la lipogénesis y la oxidación de AGs a favor de la síntesis de AGs, lo que determinaría la esteatosis hepática (Figura 2)22.
Discusión/Conclusiones
El desarrollo de la esteatosis hepática en el paciente obeso es el resultado de múltiples alteraciones metabólicas que ocurren en condiciones de desbalance dietario y que involucran la inducción de estrés oxidativo, RI, junto con niveles alterados de ciertas adipoquinas capaces de influir en la sensibilidad a la insulina (Figura 2), y que puede progresar hacia la EHNA. Se ha sugerido que la cirugía bariátrica es el procedimiento más efectivo para lograr el control del peso corporal a largo plazo en la obesidad mórbida, la cual reduce la esteatosis, la RI y las alteraciones metabólicas asociadas22 23. Un adecuado tratamiento dietético para favorecer la pérdida de peso, como un aspecto terapéutico central, podría ser combinada con la administración de:
- Antioxidantes, para minimizar o prevenir el desarrollo de estrés oxidativo, los cuales son considerados como agentes sensibilizadores a la acción de la insulina12
- AGPI n-3, con el objetivo de mejorar la eficiencia de los mecanismos de señalización en el metabolismo lipídico y RI, intervención que reduce la esteatosis hepática24, y/o
- Chaperonas químicas que mejoran las acciones sistémicas de la insulina25.
De acuerdo a estas consideraciones, estrategias terapéuticas que aumenten la sensibilidad a la insulina y las defensas antioxidantes en el hígado merecen ser evaluadas en futuros estudios controlados, con el fin de disminuir la esteatosis y evitar su progresión a la EHNA.
