Artículo de revisión
← vista completaPublicado el 28 de octubre de 2015 | http://doi.org/10.5867/medwave.2015.09.6293
Periodontitis como determinante del inicio y progresión de la enfermedad de Huntington: revisión de la literatura
Periodontitis determining the onset and progression of Huntington's disease: review of the literature
Resumen
La enfermedad de Hungtinton es un trastorno neurodegenerativo, causado por la expansión de un triplete de citosina, adenina, guanina en el gen de la huntingtina. Se manifiesta con un deterioro físico, cognitivo y psiquiátrico a distintas edades en el adulto, con un pronóstico fatal. Además del número de repeticiones del triplete, existirían otros factores que explicarían el inicio de esta enfermedad a más temprana edad. Se sabe que la neuroinflamación es un protagonista en los trastornos neurodegenerativos, no siendo la enfermedad de Huntington una excepción. La neuroinflamación exacerba el daño neuronal producido por la mutación, al existir activación aberrante de la célula microglía, disfunción de astrocitos y células dendríticas; compromiso de la barrera hematoencefálica y activación de complemento, todas ellas como efecto directo e indirecto de la mutante y otros estímulos como infecciones crónicas. Es el interés del presente trabajo analizar la periodontitis, como modelo de infección bucodental crónica y fuente de inflamación sistémica. Hipotetizamos que el potencial rol de la periodontitis en la enfermedad de Huntington y los mecanismos por los cuales contribuiría a la manifestación temprana y progreso de dicha enfermedad, para lo cual se consideraron revisiones sistemáticas, metanálisis y estudios experimentales publicados tanto en español como en inglés obtenidos del PubMed y SciELO. Son diversos los mecanismos que generan inflamación en el cerebro de estos pacientes, adquiriendo especial protagonismo los mecanismos de la inmunidad innata. Las infecciones buco dentarias crónicas, como la enfermedad periodontal, pueden constituir un factor exacerbante de la neuroinflamación que per se asocia la enfermedad de Huntington.
Introducción
La enfermedad de Huntington es un desorden neurodegenerativo primordialmente del adulto, heredado con patrón autosómico dominante, manifestándose con deterioro físico, cognitivo y psiquiátrico que es finalmente fatal [1]. La causa del síndrome es una mutación dinámica basada en una expansión específica del repetitivo citosina, adenina, guanina en el exón 1 del gen de la huntingtina. Los alelos de citosina, adenina, guanina, y más repeticiones son totalmente penetrantes y causan enfermedad de Hungtinton. Existe una relación inversa entre el tamaño del repetitivo y la edad de inicio [2]. Sin embargo, éste no sería el único determinante en las formas juveniles de la enfermedad de Huntington. Estudios recientes sobre sistema inmune y la enfermedad de Huntington [1] demuestran que el daño neuronal no sólo depende de la toxicidad de la huntingtina mutante y de sus interacciones, sino también de condiciones inflamatorias del entorno cerebral [3],[4]. En alusión a esto último, se han planteado varias teorías, algunas demostradas en modelos animales, para sustentar la existencia de inflamación en el cerebro de pacientes con enfermedad de Huntington. Las tesis más fundadas sobre la inflamación cerebral en la enfermedad de Huntington son:
- La inflamación es resultado de un proceso reactivo a la degeneración neuronal [5].
- Es producto de la acción directa de la huntingtina mutante sobre la fisiología microglial, de células dendríticas y astrocitos [1],[4],[6].
- Altera la barrera hematoencefálica [6] y activa el sistema del complemento [1].
En la literatura revisada no se encontraron datos sobre el impacto que podrían tener las infecciones crónicas en la patogénesis de enfermedad de Huntington, esto en virtud de existir algunas evidencias que le atribuyen potencial para inducir daño neuronal a través de la evolución al estadio 3 de activación microglial [1]. Frente a este antecedente, nos hemos propuesto plantear un cuarto modelo hipotético sobre el origen de la inflamación cerebral en el paciente con enfermedad de Huntington. Esta hipótesis apunta a las infecciones buco dentarias crónicas como la periodontitis y su capacidad demostrada [7],[8] para causar estados de inflamación sistémica que posteriormente conducen al transporte de citoquinas inflamatorias al cerebro, resultando en un estado de neuroinflamación persistente de bajo grado [7]. Ello contribuiría en los pacientes con enfermedad de Huntington a exacerbar el daño neuronal producido por la acción tóxica per se de los fragmentos cortos de huntingtina mutante [9]. De esta forma, se considera un determinante en el inicio y progresión de la enfermedad neuronal. Por lo tanto es un factor clave a ser tomado en cuenta, tanto por neurólogos como dentistas, en el manejo de los pacientes con antecedentes familiares o que portan la mutación causante de enfermedad de Huntington.
Esta revisión tiene como objetivos el exponer el efecto de la neuroinflamación en la patogénesis de la enfermedad de Huntington, junto con plantear un modelo hipotético que explique los mecanismos moleculares por los cuales la periodontitis, como modelo de infección buco dentaria crónica, produce neuroinflamación e indirectamente neurodegeneración.
Material y métodos
Para esta revisión recurrimos a las bases de datos PubMed y SciELO, entre el 1 de julio y el 1 de agosto de 2015. Se seleccionaron estudios experimentales, revisiones sistemáticas y metanálisis publicados entre los años 2000 y 2015, tanto en español como en inglés. Se usaron como términos de búsqueda: enfermedad de Huntington, periodontitis, neuroinflamación, neurodegeneración e inflamación sistémica.
Resultados
Neuroinflamación en la enfermedad de Huntington
Con respecto al primer objetivo planteado, la información revisada nos habilita para señalar que en la enfermedad de Huntington es característica la exacerbación y presencia de distintos elementos inmunes como consecuencia de la acumulación de huntingtina mutante en las células de glía y en macrófagos. Estos últimos son responsables de la activación autónoma de la respuesta inmune en el cerebro [10]. Por razones didácticas, se procederá a explicar detalladamente uno a uno, pero es necesario comprender su accionar en conjunto y no como procesos independientes.
Células gliales: microglía y astrocitos
Se ha observado en pacientes con enfermedad de Huntington una importante acumulación de microglía y astrocitos activados, que puede apreciarse en aquellos individuos con el gen defectuoso hasta 15 años antes de la edad estimada para la manifestación de la enfermedad [10]. En la microglía, el trabajo de Crotti et al., correlacionó la acumulación de huntingtina mutante con un aumento en la expresión y en la actividad transcripcional de los factores determinantes del linaje mieloide, PU.1 y C/EBP. Éstos son responsables, además del desarrollo y función de los macrófagos y la microglía, de determinar la actividad regulatoria de las mismas a través de la selección de potenciadores y promotores dependientes de distintos factores de transcripción como el factor de transcripción nuclear β [4], el cual es responsable del aumento de citoquinas y quimioquinas (Figura 1). Se ilustra como la expresión de huntingtina mutante en la microglía produce polarización a fenotipo M1 proinflamatorio en esta célula, con la consecuente liberación de citoquinas y especies reactivas del oxígeno, lo cual genera daño neuronal y astrogliosis. Las neuronas lesionadas liberan patrones moleculares asociados a daño, potenciando así la activación de la célula de microglía, y por tanto la neuroinflamación y sus consecuencias sobre las células de glía [1].
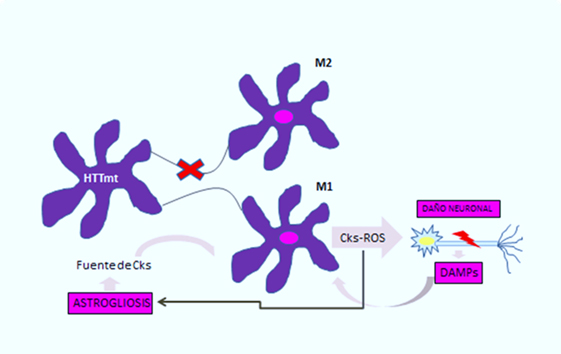
Figura 1: Círculo vicioso de activación microglial en la enfermedad de Huntington.
En el caso de los astrocitos, modelos murinos demuestran que la acumulación de huntingtina mutante reduce la expresión en membrana de un canal de potasio (Kir4.1), generando despolarización e incremento en los niveles extracelulares del ion K+, lo cual afecta la remoción de glutamato en el espacio extracelular y causa excitotoxicidad en el tejido neuronal [11],[12] (Figura 2). Esta disminución en la neuroprotección proporcionada por los astrocitos, se correlaciona con el rol funcional de la neuroinflamación en la enfermedad de Huntington [1]. A esto se suma la astrogliosis (hipertrofia y aumento en la proliferación de astrocitos) que sufren estas células como consecuencia de los mediadores proinflamatorios secretados por la microglia reactiva, y la activación aberrante de factor de transcripción nuclear β, exacerbando la respuesta inflamatoria y favoreciendo el proceso de neurodegeneración, [3] al secretar más citoquinas proinflamatorias que activarían a más células de microglía generando un círculo vicioso y progresivo [13] (Figura 1). En la Figura 2 se ilustra como la acumulación de huntingtina mutante en los astrocitos generaría un down-regulation (disminución) de un canal de potasio (Kir4.1), teniendo como consecuencia una despolarización de membrana en los mismos, esto incrementa los niveles extracelulares de K+, afectando la remoción de glutamato del espacio extracelular y generando así excitotoxicidad en el tejido neuronal [12].
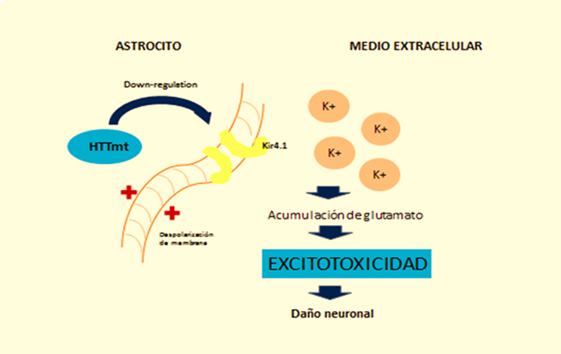
Figura 2: Efecto de la huntingtina mutante sobre la fisiología del astrocito.
Macrófagos
Así como la microglía, el fenotipo de los macrófagos puede ser de tipo M1, proinflamatorio, o de tipo M2, antiinflamatorio, cambiando entre uno u otro en función del estadio de la enfermedad [14].
Los macrófagos son los responsables de la producción de citoquinas a nivel periférico, las cuales atraviesan la barrera hematoencefálica y activan a la microglía. Su accionar se ve alterado en diversas enfermedades neurodegenerativas como la enfermedad de Huntington, ya que se ha descrito una alteración en la respuesta de los macrófagos a quimioatractantes y consecuentemente una migración reducida hacia focos de inflamación en pacientes con esta patología. Ello se correlacionaría con un aumento en la secreción de citoquinas proinflamatorias. Se presume que la causa de esto es la acumulación de huntingtina mutante que utilizaría los mismos mecanismos que en la microglía para interferir con el funcionamiento normal de estas células [1]. También habría una polarización hacia el fenotipo M1, generando una significativa reducción en los niveles del factor de crecimiento transformante β1, el cual es producido por el fenotipo M2 y tiene propiedades neuroprotectoras, participando en la regulación de la activación microglial. Por lo que sus niveles plasmáticos, son también predictivos para determinar la edad de debut de la patología [14].
Sistema de complemento
Siendo uno de los elementos más importantes del sistema inmune innato y un punto de conexión entre éste y el sistema inmune adaptativo, es de particular interés su rol en la enfermedad de Huntington. En el sistema nervioso central, la mayoría de los componentes y receptores del mismo son expresados tanto en células de glía como en neuronas y puede ser activado por distintas proteínas, entre ellas la huntingtina mutante. Esto gatilla una cascada de procesos como la secreción de citoquinas proinflamatorias, atracción de macrófagos, aumento en la fagocitosis de antígenos y lisis celular, entre otras [1].
Citoquinas y quimioquinas
Como ya se ha mencionado, los niveles de citoquinas y quimioquinas proinflamatorias se encuentran aumentados en la enfermedad de Huntington y son las responsables de la activación de la microglía y de la astrogliosis, así como también de la migración leucocitaria desde la periferia y de la disfunción de la barrera hematoencefálica [14]. Son de particular interés la IL-6, la IL-1β, el TNF-α [1] y cinco quimioquinas (eotaxin-3, MIP-1β, eotaxin, MCP-1 y MCP-4) [15]. Cabe remarcar que la IL-1β, secretada por varios tipos celulares como las células dendríticas, induce directamente la neurotoxicidad por la activación de vías de señalización dependientes de tirosinquinasas y la fosforilación de receptores de n-metil-D-aspartato involucrados en la vía de factor de transcripción nuclear β [1]. Se ha encontrado altos niveles de IL-1β y TNF-α en enfermedades neurodegenerativas, alterando la función neuronal y afectando la concentración de neurotransmisores como el glutamato [16], que como ya hemos mencionado, su exceso conduce a neurotoxicidad [11],[12]. También se ha observado in vitro que la combinación IL-1β más TNF-α, genera muerte neuronal y apoptosis [16].
Rol del sistema inmune adaptativo
Si bien la respuesta inmune implica una interacción balanceada entre el sistema inmune innato y el sistema inmune adaptativo, hasta la fecha según Ellrichmann et al. no se ha podido establecer el rol del sistema inmune adaptativo en la enfermedad de Huntington, pese a que se sugiere que las células dendríticas, los linfocitos T y determinadas citoquinas específicas de este sistema; podrían estar involucradas ya que la acumulación de huntingtina mutante en las células dendríticas ocasionaría su constante activación. Éstas a su vez, activarían a los linfocitos T, los que segregarían más citoquinas agravando el cuadro inflamatorio [1].
Inflamación y la barrera hematoencefálica
La inflamación crónica a nivel periférico y/o intersticial afectaría la pared capilar de la barrera hematoencefálica, permitiendo el ingreso al sistema nervioso central de péptido amiloide [7], de células inflamatorias y proteínas séricas como citoquinas y quimioquinas [17], agravando y complementando lo anteriormente expuesto.
Neuroinflamación y formación de proteínas amiloides
Se ha reportado que la neuroinflamación induciría hiperfosforilación de proteína TAU [7] con la consecuente formación de intermediarios oligómeros y ovillos neurofibrilares, que en los pacientes con enfermedad de Huntington, interactuarían con los fragmentos de huntingtina mutante formando agregados proteicos que producen disfunción neuronal y neurodegeneración [9]. Adicionalmente, la inflamación cerebral puede aumentar la amiloidogénesis elevando la proteólisis del péptido precursor de amiloide, resultando en un incremento en la concentración de monómeros o dímeros de amiloide. Éstos, en función del tiempo, formarían intermediarios oligómeros que han demostrado en modelos animales ser neurotóxicos, llevando a la degeneración neuronal por activación de vías apoptóticas de muerte [18].
Periodontitis crónica: una enfermedad inflamatoria sistémica de bajo grado
Con respecto al segundo objetivo planteado, la información revisada define a la periodontitis como una enfermedad inflamatoria destructiva, polimicrobiana, que compromete los tejidos de protección e inserción dental (tejido conectivo gingival adherido a la superficie radicular, ligamento periodontal, cemento radicular y hueso alveolar adyacente) [8]. Clínicamente se caracteriza por sangrado y secreción purulenta de las encías, profundización progresiva del surco gingival (referido como bolsa periodontal), halitosis oral, diastemas y movilidad dental en estadios avanzados [7]. Es considerada la primera causa de pérdida dental [8].
La placa dental es la principal fuente de periodontitis, consistiendo predominantemente en bacterias Gram negativas y anaerobias. Sin embargo, el factor microbiano no es suficiente para mediar la destrucción progresiva del tejido periodontal, siendo imprescindible para tal efecto, la respuesta del huésped, que en última instancia será el que determine la intensidad de la destrucción tisular. La forma en cómo responda el huésped al ataque de los periodonto-patógenos estará condicionado por factores de riesgo genéticos y ambientales o adquiridos [7].
El revestimiento ulcerado de la bolsa periodontal constituye una puerta de entrada a las bacterias y sus productos nocivos en la circulación sistémica. Se ha reportado que el área total de superficie del revestimiento ulcerado de la bolsa periodontal en pacientes con periodontitis severa es aproximadamente de 15 a 20 centímetros cuadrados [7], lo que permite el ingreso de bacterias y sus productos nocivos a la circulación sistémica por medio de tres mecanismos [19]:
- Infección metastásica o bacteriemia: los microorganismos que ingresan al torrente sanguíneo, no son eliminados y se diseminan.
- Daño metastásico: por las endotoxinas y lipopolisacáridos liberados y letales para las células.
- Inflamación metastásica: por las reacciones del antígeno anticuerpo y la liberación de mediadores químicos.
Esto altera el carácter de la periodontitis de una enfermedad local a un desorden sistémico, capaz de sostener una “inflamación sistémica de bajo grado” [7].
Vínculo entre periodontits y enfermedad de Huntington
La periodontitis como enfermedad sistémica de bajo grado, puede ser un determinante en el inicio y progresión de la enfermedad de Huntington, a través de dos mecanismos potenciales (Figura 3):
- Inflamación sistémica precedida por periodontitis.
- Influencia bacteriana y viral directa.
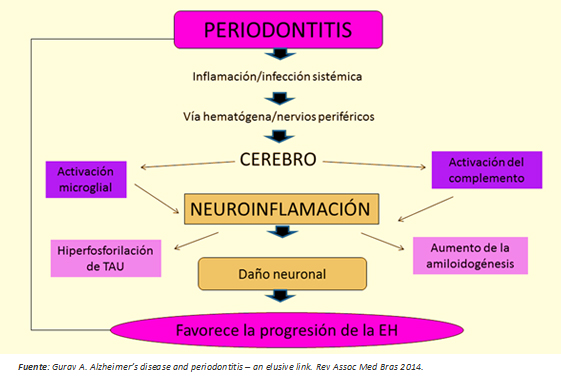
Figura 3: Nexo entre periodontitis y enfermedad de Huntington.
Con respecto al primer mecanismo, las bacterias y la respuesta del huésped elevan los niveles séricos de citoquinas proinflamatorias [7],[8], estableciéndose un estado de inflamación sistémica que compromete la integridad de la barrera hematoencefálica. Previamente, ésta estaría alterada per se en la enfermedad de Huntington [6], facilitando el acceso de estas moléculas al cerebro. Ellas activarían a la célula de microglía, llevándola al estadio 3 de maduración [1],[10], adoptando de esta forma un fenotipo proinflamatorio M1 con liberación de citoquinas tales como IL-1B, TNF-a, IL-6, y otras moléculas inflamógenas que activarían el sistema del complemento a nivel cerebral [8]. Este proceso genera un estado de “neuroinflamación persistente de bajo grado”, con capacidad de causar muerte neuronal por dos caminos: activación de vías apoptóticas [16] y aumento en la concentración del neurotransmisor glutamato produciendo neurotoxicidad [11],[12].
El segundo mecanismo involucra la invasión cerebral por bacterias y virus residentes de la placa dental, los cuales serían transportados o por vía sanguínea o por vía de nervios periféricos [7]. Las investigaciones que avalan este segundo mecanismo propuesto son:
- Riviere et al., aislaron especies espiroquetales tales como Treponema denticola, Treponema pectinovorum, Treponema vincentii, Treponema amylovorum, Treponema maltophilum, Treponema medium y Treponema socranskii, del cerebro de sujetos con enfermedad de Alzheimer, utilizando PCR (reacción en cadena de la polimerasa) específica. Se ha especulado que las especies treponémicas de cavidad oral pueden tener acceso a la corteza cerebral vía nervio trigeminal [20]. A este respecto, un estudio encontró una asociación estadísticamente significativa entre espiroquetas y enfermedad de Alzheimer, en función de detectar treponemas en el 93,7% de los pacientes con enfermedad de Alzheimer frente al 33,3% de los sujetos control [21].
- Poole et al., detectaron componentes de Pophyromonas gingivalis en el cerebro de sujetos con enfermedad de Alzheimer [22]. A partir de este hallazgo se ha planteado la asociación de enfermedad periodontal con la edad de inicio y el perpetuamiento de la inflamación en enfermedad de Alzheimer. Se ha demostrado que el Pophyromonas gingivalis es un experto en evadir la respuesta inmune del huésped, usando para ello varios mecanismos como:
a) Capacidad natural de formar biofilm [23].
b) Degradar componentes del complemento a través de la producción de proteasas [24].
c) Reclutando proteínas regulatorias del huésped como factor H y proteína C4 [25].
d) Capacidad de adherirse a eritrocitos vía receptor 1 del complemento (CR1), éste le permite a la bacteria
pasar indetectable para los fagocitos circulantes, y de ser transportada vía circulación sistémica
accediendo a órganos remotos del cuerpo en individuos con susceptibilidad para desarrollar patologías
inflamatorias [26].
- Singhrao et al., consideran a la Pophyromonas gingivalis como un patógeno inflamofílico clave, capaz de establecer comunidades con otros microbios, exacerbando la inflamación en el huésped. También proponen que el ingreso de patógenos periodontales al cerebro produce inflamación a través de la activación microglial y del sistema del complemento [8].
En resumen, la periodontitis crónica sería una fuente potencial de daño neuronal a través de su capacidad probada para generar inflamación sistémica de bajo grado y diseminación al cerebro de patógenos periodontales vía hematógena y neural. Ambas situaciones activan predominantemente el sistema inmune innato, mediante la polarización del fenotipo M1 de la célula de microglía y por la activación por vía alterna del sistema de complemento, que en última instancia, potencia la activación microglial con la consecuente liberación de citoquinas y especies reactivas del oxígeno (Figura 4). Según Ellrichmann et al., ello produciría un fenómeno de astrogliosis (hipertrofia y aumento en la proliferación de astrocitos como respuesta a estímulos proinflamatorios) [3]. Con ello se exacerba la inflamación cerebral promovida, tanto por la inflamación sistémica como por la misma enfermedad de Huntington. Esta neuroinflamación produce lesión neuronal por:
- Activación de vías apoptóticas.
- Hiperfosforilación de proteína TAU.
- Aumento de la amiloidogénesis.
Las neuronas lesionadas liberarían patrones moleculares asociados a daño en etapas tardías del proceso de muerte, potenciando la activación microglial en el cerebro, y por tanto el daño neuronal, conduciendo al deterioro cognitivo [27] (Figuras 1 y 3).
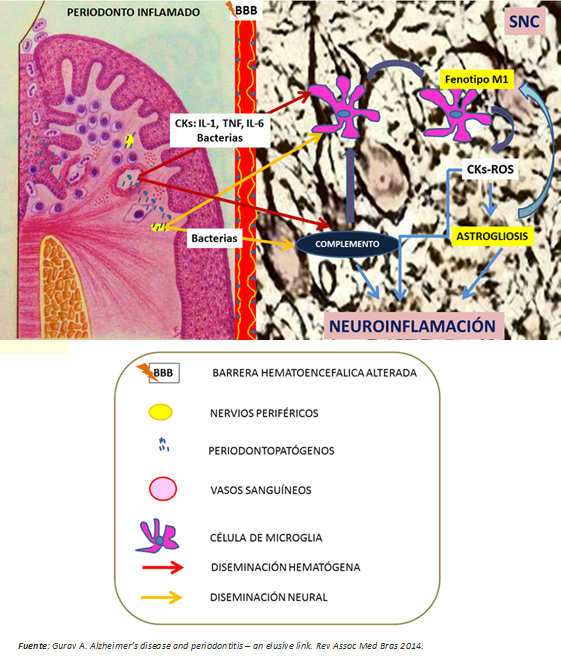
Figura 4: Nexo entre periodontitis y enfermedad de Huntington.
Consideraciones finales
Gracias al desarrollo en la biología molecular se pudo identificar la causa de enfermedad de Huntington. Sin embargo, aún se desconoce el mecanismo exacto que produce el daño neuronal y el deterioro cognitivo en estos pacientes. Recientes reportes le otorgan a la neuroinflamación un rol clave en la patogénesis de la enfermedad de Huntington [4],[6]. Todo el conocimiento del que se dispone actualmente sobre este tema es resultado de modelos animales, los cuales le atribuyen a la huntingtina mutante la responsabilidad de promover y orquestar el escenario inflamatorio en el cerebro de estos pacientes, a través de mecanismos de inmunidad innata y adaptativa [1]. Pero además, esta neuroinflamación orquestada por huntingtina mutante, puede verse exacerbada por infecciones sistémicas con capacidad de generar y mantener un estado inflamatorio periférico de bajo grado.
Nosotros planteamos que las infecciones buco dentarias crónicas como la enfermedad periodontal, podrían constituir un determinante en el inicio y progresión de la enfermedad de Huntington, por los mecanismos que se han revisado. No obstante, recalcamos que no existen modelos animales que confirmen nuestra hipótesis, por lo que este tema debería ser tomado en cuenta para futuras investigaciones sobre esta enfermedad. De demostrarse esta asociación, se permitirá un abordaje multidisciplinario de los pacientes con enfermedad de Huntington y aquellos con antecedentes familiares o que portan la mutación causante de la enfermedad, ya que el diagnóstico precoz de patologías bucales infecciosas con impacto sistémico en este grupos de pacientes permitiría controlar el grado de deterioro cognitivo. Así se prevendría un debut temprano y un curso evolutivo fatal de la enfermedad neuronal.
Conclusiones
Son diversos los mecanismos que generan inflamación en el cerebro de pacientes con enfermedad de Huntington, adquiriendo especial protagonismo los mecanismos de la inmunidad innata. Las infecciones buco dentarias crónicas como la periodontitis, pueden constituir un factor exacerbante de la inflamación que en sí misma acompaña a esta enfermedad.
Ante este antecedente, sugerimos se investigue en modelos clínicos y preclínicos la asociación de periodontitis y enfermedad de Huntington para así poder retrasar el inicio de dicha patología a partir de la implementación de cuidados adicionales y medidas de prevención desde el lugar del odontólogo.
Este mismo modelo que proponemos, podría expandirse a otras afecciones inflamatorias crónicas comunes. Una vez comprobadas, permitiría actuar en consecuencia mejorando así la calidad de vida del paciente.
Notas
Las autoras nos hacemos responsables de los contenidos y la originalidad de las ilustraciones del presente trabajo, y declaramos no tener conflictos de intereses.
Conflictos de intereses
Las autoras han completado el formulario de declaración de conflictos de intereses del ICMJE, y declaran no haber recibido financiamiento para la realización del artículo y no tener otros conflictos de intereses con la materia del artículo. Los formularios pueden solicitarse al autor o la Revista.
