Reporte de caso
← vista completaPublicado el 4 de junio de 2019 | http://doi.org/10.5867/medwave.2019.05.7645
Síndrome miasténico congénito por deficiencia de rapsina: reporte de caso con nueva mutación y heterocigosidad compuesta
Congenital myasthenic syndrome due to rapsyn deficiency: A case report with a new mutation and compound heterozygosity
Resumen
Introducción Los síndromes miasténicos congénitos son un grupo heterogéneo de desórdenes genéticos, caracterizados por una transmisión sináptica anormal en la placa neuromuscular.
Reporte Presentamos el caso de un paciente de dos años, varón, con hipotonía, ptosis palpebral y debilidad simétrica y de predominio proximal, caracte-rísticas que aparecieron desde el nacimiento y que motivaron varias hospitalizaciones por neumonía e insuficiencia ventilatoria. Desde el inicio de la deambulación a los dos años, los padres notaron que la debilidad empeoraba por las tardes y con la actividad física repetida o prolongada. El examen físico a los dos años mostró ptosis palpebral, debilidad de predominio proximal y fatigabilidad con el esfuerzo sostenido. La electro-miografía evidenció decremento del 27% en el potencial de acción muscular compuesto. El análisis de tríos mostró heterocigosis compuesta por transmisión de dos mutaciones diferentes en el gen de rapsina, una ya conocida procedente del padre y la otra no reportada previa-mente, procedente de la madre. El paciente recibió piridostigmina obteniendo mejoría inmediata y logrando un desempeño óptimo en activi-dades escolares, deportivas y de la vida cotidiana. A la fecha, no ha presentado nuevos episodios de insuficiencia ventilatoria.
Conclusiones La debilidad de inicio neonatal y la fatigabilidad o agotamiento con el esfuerzo sostenido, con afección principalmente de los músculos con inervación troncal y con un decremento mayor al 10% en el potencial de acción muscular compuesto en la electromiografía, deben hacer sospechar en un síndrome miasténico congénito. Se revisan los puntos clínicos clave que permiten establecer el diagnóstico oportuno y las opciones de tratamiento efectivo para algunos de estos síndromes.
|
Ideas clave
|
Introducción
Los síndromes miasténicos congénitos son un grupo heterogéneo de desórdenes genéticos, causados por una transmisión anormal en la placa neuromuscular[1]. La mayoría derivan de defectos moleculares en el receptor de acetilcolina del músculo, pero también pueden ser causados por otras alteraciones genéticas[1],[2]. Es importante diferenciarlos de la miastenia neonatal, en la cual las madres con miastenia gravis transmiten anticuerpos contra el receptor de acetilcolina por la placenta. Por otro lado, también debe diferenciarse de la miastenia gravis adquirida en la cual el mecanismo fisiopatológico es el fenómeno autoinmune y que es muy rara antes de los dos años de edad.
El caso que presentamos a continuación resalta la importancia de la detección clínica del fenómeno de fatigabilidad o agotamiento, para establecer la sospecha y evitar la demora en el diagnóstico que puede amenazar la vida del paciente. Es necesario resaltar que algunos síndromes miasténicos congénitos cuentan con tratamiento efectivo que mejora la calidad de vida y el pronóstico[3],[4].
Reporte de caso
Anamnesis
Paciente varón de dos años seis meses, sin antecedentes prenatales de importancia, nació por cesárea programada sin complicaciones y desde el nacimiento presentó marcada hipotonía y debilidad global, caracterizada por ausencia de expresión facial y movimientos corporales, severo trastorno de succión-deglución que impedía la alimentación directa e hipoventilación asociada a un episodio de neumonía y sepsis requiriendo ventilación mecánica por 45 días. El egreso hospitalario fue a los dos meses con indicación de terapia física, terapia de succión-deglución y alimentación por sonda nasogástrica. La succión fue mejorando lentamente y a los ocho meses logró alimentarse en forma directa dejando la sonda. En cuanto al desarrollo motor, sostuvo la cabeza a los ocho meses, se sentó a los 10 meses y a los dos años logró caminar por trechos cortos. Desde los primeros meses de vida, los padres notaron que la ptosis palpebral y la debilidad facial empeoraban por las tardes. Desde los dos años, cuando empezó a caminar, advirtieron que tenía mayor dificultad por las tardes, dificultando su desplazamiento y las actividades recreativas. En cuanto al desarrollo del lenguaje, inició balbuceos a los ocho meses y a los dos años con cinco meses logró elaborar frases cortas. El desarrollo social fue normal.
A los siete meses tuvo un episodio de neumonía aspirativa requiriendo ventilación mecánica invasiva por 12 días, saliendo de alta luego de un mes. A los 11 y a los 15 meses volvió a hospitalizarse por neumonía requiriendo ventilación mecánica por 10 días y cuatro días respectivamente. Salió de alta con diagnóstico de neumonía recurrente e hipotonía muscular. Desde la segunda hospitalización usaba β-2 agonistas inhalatorios cuando tenía síntomas respiratorios notando mejoría con este tratamiento.
A los 23 meses se le realizó orquidopexia por criptorquidia bilateral.
El padre de 32 años, la madre de 30 años y el hermano de dos meses no presentaban debilidad ni otros problemas de salud.
Examen físico
Funciones vitales: frecuencia cardiaca: 96 por minuto, frecuencia respiratoria: 18 por minuto. Peso, talla y perímetro cefálico adecuados para la edad.
Lucía en regular estado general, de contextura delgada, colaborador con el examen y el interrogatorio. Desde el ingreso al consultorio llamó la atención su facies miopática y poco expresiva, con ptosis palpebral bilateral, cara alargada, cejas arqueadas, hipoplasia medio facial, boca entreabierta con labio inferior evertido, paladar ojival, diastema y mentón en punta (Figura 1).
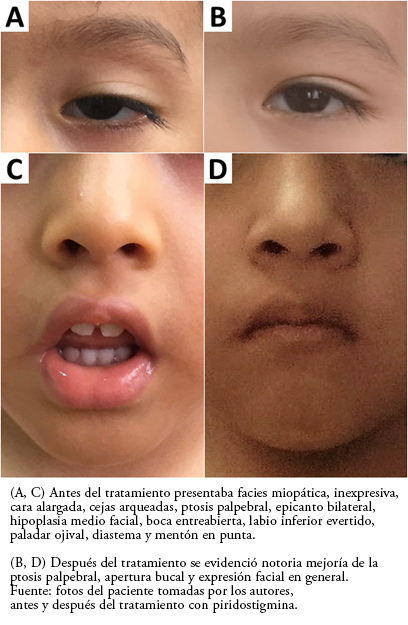 Tamaño completo
Tamaño completo El examen neurológico mostró memoria normal, lenguaje con comprensión adecuada y discreta disartria. Se evidenció debilidad muscular de predominio proximal (Figura 2A) con signo de Gowers positivo, hipotonía generalizada y sensibilidad normal. Los reflejos osteotendinosos iniciales fueron normales (Figura 2B) aunque la percusión repetida durante dos minutos mostró evidente agotamiento del reflejo bicipital. Se encontró discreto hipotrofismo muscular. La sensibilidad táctil, dolorosa, térmica y propioceptiva fue normal. El resto del examen no fue contributario. La mirada hacia arriba sostenida con estímulo permanente por dos minutos (vídeo animado infantil) mostró agotamiento en la apertura palpebral.
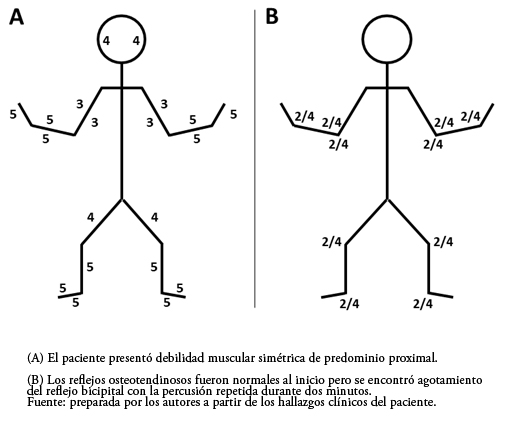 Tamaño completo
Tamaño completo Inmediatamente se estableció el diagnóstico de síndrome miasténico congénito basándonos principalmente en la debilidad de predominio proximal, la facies miopática, el fenómeno de agotamiento en el transcurso del día y el inicio de la enfermedad en la etapa neonatal. En el diagnóstico diferencial se consideró a la distrofia muscular de cinturas y algunas miopatías congénitas. Se solicitó anticuerpos contra el receptor de acetilcolina y anticuerpos contra la tirosina kinasa específica de músculo, creatinfosfokinasa, gases arteriales, electrolitos, lactato, piruvato y transaminasas resultando normales todas estas pruebas. La electromiografía mostró decremento del 27% en el potencial de acción muscular compuesto a la estimulación repetitiva del nervio facial. Se recetó piridostigmina a siete milígramos por kilo al día y terapia física.
En el control a las dos semanas los padres comentaron mejoría inmediata con el uso de piridostigmina, tanto en la forma de caminar (logrando, incluso, correr) como en la apertura palpebral, la articulación de la palabra y la postura al sentarse. El examen mostró mejoría de la fuerza y el tono muscular, desapareciendo el signo de Gowers. El tratamiento con piridostigmina a siete milígramos por kilo al día en cinco tomas diarias, se mantiene hasta la actualidad.
Se le realizó una resonancia magnética de cerebro, cuyo primer informe indicó “agenesia de cuerpo calloso”. Sin embargo, una observación más detallada por radiólogos y neurólogos corrigió dicho informe, pues el cuerpo calloso y todo el encéfalo eran totalmente normales.
Estudio genético
Se enviaron muestras del paciente, de sus dos progenitores y del hermano menor al Laboratorio de Investigación Neuromuscular de la Clínica Mayo, de Rochester, Minnesota, para la investigación de síndromes miasténicos congénitos mediante el estudio de genoma completo (con estudio de tríos de caso-progenitores). En dicho estudio se demostró que el probando (II.1) tiene dos mutaciones puntuales en heterocigosis en el exón 2 del gen de la rapsina. La primera consiste en una mutación puntual de tipo transversión que sustituye una citosina por una adenina en la posición 264 del ácido desoxirribonucleico (ADN), ocasionando el cambio a nivel de proteína de la asparagina por una lisina en la posición 88 (p.N88K). La segunda es otra mutación puntual de tipo transversión que sustituye una citosina por una adenina en la posición 218 del ADN, produciendo el cambio a nivel de proteína de la alanina por un aspartato en la posición 73 (p.A73D). Esto hace que el paciente sea un heterocigoto compuesto en el gen RAPSN. Se encontró que el padre (I.1) es portador de la mutación missense RAPSN N88K en heterocigosis la cual ha sido descrita previamente como variante patogénica. Tanto la madre (I.2) como el hermano menor (II.2) son portadores de la mutación A73D en heterocigosis (Figura 3). Esta segunda mutación no se encuentra descrita previamente en las bases genéticas consultadas (SNP databases y Clinvar), pero el análisis de esta mutación con los programas informáticos predictores de efecto PolyPhen-2 y Provean/Sift, la clasifica como variante patogénica (causante de enfermedad).
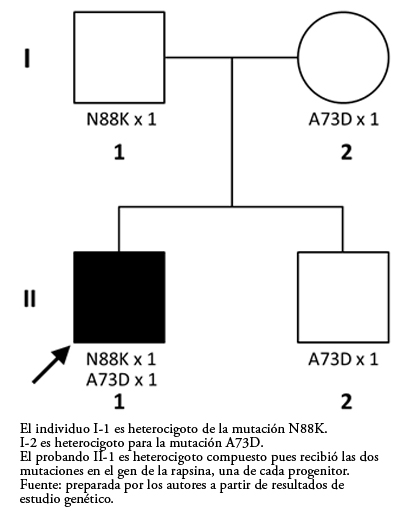 Tamaño completo
Tamaño completo Discusión
Los síndromes miasténicos congénitos son un grupo heterogéneo de alteraciones genéticas que tienen como consecuencia una transmisión sináptica anormal entre las terminales axónicas motoras y cada fibra de músculo esquelético[1],[2]. En conjunto alcanzan una prevalencia que varía entre 2,8 a 15,5 por 1 000 000 de habitantes, aunque podría estar subestimada[3],[5].
La mayoría de estos casos derivan de defectos moleculares en el receptor nicotínico de acetilcolina del músculo, pero también pueden ser causados por mutaciones en proteínas presinápticas, mutaciones en proteínas asociadas a la lámina basal sináptica, defectos en el desarrollo o el mantenimiento de la placa mioneural o defectos en la glicosilación de proteínas[1],[2].
En cuanto a la sintomatología, se caracterizan por miastenia o “fatigabilidad” del músculo esquelético, con inicio desde del nacimiento o en los primeros meses de vida, tal como ocurrió con nuestro paciente. Rara vez, los síntomas aparecen en la segunda o tercera década. Es importante mencionar que la gravedad y el curso de la enfermedad son muy variables, desde síntomas menores hasta debilidad progresiva. En algunos tipos de síndromes miasténicos congénitos, los síntomas de debilidad pueden ser leves, aunque pueden ocurrir exacerbaciones repentinas de agotamiento o incluso episodios agudos de insuficiencia respiratoria desencadenados por fiebre, infecciones o estrés. En el inicio neonatal es frecuente la insuficiencia respiratoria, episodios súbitos de apnea, dificultades de alimentación y llanto débil. También puede encontrarse artrogriposis múltiple congénita y estridor. Cuando el debut es más tardío, la debilidad muscular que se acentúa con la actividad física como correr o subir escaleras y que empeora a lo largo del día es la característica principal. La ptosis palpebral y la oftalmoparesia fija o fluctuante son formas comunes de presentación. El músculo cardíaco y el músculo liso no suelen estar involucrados3,5,6. En la bibliografía revisada sobre el gen RAPSN no se ha asociado dentro del cuadro clínico a la criptorquidia por lo que podría ser un problema de salud aislado sin relación con las alteraciones genéticas encontradas. No se ha descrito en la literatura médica, diferencias en la frecuencia de presentación ni en la expresión clínica de los síndromes miasténicos congénitos en cuanto al género.
Creemos que en nuestro paciente la facies miopática, la ptosis palpebral y la criptorquidia, notorias al nacimiento, contribuyeron al etiquetado genérico de “síndrome dismórfico” que llevó a buscar una malformación del sistema nervioso. Posteriormente, una resonancia magnética de mala calidad y probablemente una apreciación poco cuidadosa o de cortes incompletos, llevó al diagnóstico erróneo de “agenesia del cuerpo calloso” que también constituyó un distractor.
En la evolución, durante las sucesivas atenciones en la emergencia por enfermedad respiratoria, debió prestarse más atención a la facies miopática y al fenómeno de agotamiento con el transcurrir del día. Los problemas de deglución, la aspiración recurrente, las neumonías a repetición y la necesidad de ventilación mecánica prolongada, siempre son sugerentes de enfermedades neuromusculares.
La estimulación nerviosa repetitiva estudia la variación de la amplitud del potencial de acción muscular compuesto, obtenido en respuesta a sucesivos estímulos eléctricos sobre el nervio. Normalmente, la disminución de amplitud entre el primer y cuarto potencial de acción muscular compuesto no debe ser mayor al 8%. Cuando el decremento a la estimulación repetida a baja frecuencia es mayor al 10%, como ocurrió en este paciente, debe sospecharse de miastenia gravis, síndrome de Lambert Eaton o un síndrome miasténico congénito1.
Cuando se tiene la sospecha de un síndrome miasténico congénito, es importante buscar el diagnóstico específico a través de paneles de estudios genéticos, como se hizo con nuestro paciente. La utilidad de los paneles genéticos radica en que existen muchos genes cuyas alteraciones pueden dar una presentación clínica muy similar, por lo que estos estudios abaratan los costos. Por otro lado, identificar la mutación causal permite en algunos casos, no solo la administración del tratamiento más adecuado, sino también establecer el pronóstico y la adecuada consejería genética a la familia[6].
El fenotipo descrito en nuestro paciente es coincidente al descrito en pacientes con mutaciones en el gen RAPSN (proteína de la sinapsis asociada al receptor) que codifica a la proteína rapsina que conecta y estabiliza los receptores de acetilcolina a la membrana postsináptica y los acopla al citoesqueleto subsináptico[7],[8]. Se ha descrito que las mutaciones en este gen causan un síndrome miasténico congénito postsináptico caracterizado por deficiencia de receptores de acetilcolina en la placa mioneural[7],[8],[9],[10].
La mayoría de los pacientes presentan un cuadro clínico de inicio temprano (usualmente en etapa neonatal), con síntomas miasténicos tales como ptosis palpebral bilateral, debilidad facial y de otros músculos troncales, así como debilidad en los músculos de extremidades superiores e inferiores. El fenómeno de fatigabilidad a lo largo del día es característico y debe investigarse siempre en el interrogatorio. Es común encontrar en la anamnesis la ocurrencia de episodios de debilidad severa súbita provocada por estrés, fiebre o infecciones, a menudo acompañados de insuficiencia respiratoria que puede generar la necesidad de ventilación mecánica como ocurrió en nuestro paciente. Otras manifestaciones clínicas son la hipotonía, las apneas episódicas y los trastornos de succión y deglución. El cuadro clínico puede variar de leve a severo[3],[7],[7],[10].
Existen cerca de 200 variantes patogénicas identificadas en gen RAPSN. La mutación missense c.264C > A (p.N88K) ha sido identificada en muchos individuos como variante patogénica en la región codificante en por lo menos un alelo, encontrándose evidencia de un origen indoeuropeo; aunque no todos los individuos que la presentan tienen el mismo haplotipo[3],[7]. Se han descrito casos de homocigosidad, heterocigosidad compuesta y deleciones. También se han identificado pacientes con variantes patogénicas en la región del promotor[1],[8],[10],[11].
La mutación A73D encontrada tanto en el paciente como en su madre y hermano menor no ha sido descrita hasta el momento. Como se ha mencionado, el análisis según los programas informáticos predictores de efecto, las bases de variación genética de polimorfismo de nucleótidos únicos y las bases de datos acerca de la variación genómica y su relación con la salud humana utilizados la clasifican como variante patogénica (es decir, causante de enfermedad). Al asociarse en el paciente con la mutación N88K proveniente del padre resultan en la aparición de enfermedad que se expresa en el paciente con las manifestaciones clínicas de un síndrome miasténico congénito, esta nueva mutación patogénica A73D, debería rastrearse en la población empezando por estudios genéticos complementarios en otros integrantes de la familia amplia y buscando variaciones en la presentación clínica.
En cuanto al tratamiento, es necesario resaltar que los pacientes con RAPSN N88K presentan una respuesta favorable a la medicación con inhibidores de la enzima anticolinesterasa[3],[7],[8],[12]. Esto coincide con nuestro paciente que tuvo gran mejoría con la piridostigmina. En dos estudios de seguimiento a largo plazo de pacientes con síndrome miasténico congénito por deficiencia de rapsina el uso de piridostigmina tuvo una excelente respuesta. En pacientes con respuesta parcial puede usarse 3,4-diaminopiridina que produce mejoría adicional y permite reducir la dosis de piridostigmina. También se ha descrito mejoría en los pacientes con deficiencia de rapsina con el uso de salbutamol, lo cual también ocurrió en nuestro paciente[1],[5],[8],[12]. Es importante recalcar que la mejoría con la piridostigmina ocurre en los pacientes con deficiencia de rapsina, independientemente de la mutación genética que la ocasiona y del tipo de herencia que la provoca. Ello se debe a un retraso en la hidrólisis de la acetilcolina facilitando la interacción con los receptores y la transmisión sináptica.
El pronóstico a largo plazo es bueno. Por lo general, el curso clínico es estable excepto por la debilidad intermitente que puede desencadenarse por infecciones menores o fiebre y puede ocurrir a lo largo de toda la vida a pesar de la medicación. Las crisis respiratorias disminuyen tanto en frecuencia como en severidad y son muy raras después de los seis años de edad. Por el contrario, las exacerbaciones caracterizadas por debilidad cervical y de cinturas, síntomas bulbares y ptosis continúa observándose hasta la adultez[12]. Es necesario el seguimiento a largo plazo de estos pacientes para afinar el pronóstico relacionándolo a las mutaciones presentadas.
Conclusiones
Queremos enfatizar que la sospecha de un síndrome miasténico congénito debe hacerse en todo paciente que acuda con historia de fatiga muscular (debilidad que empeora con el transcurrir del día), con afección especialmente de los músculos de la cara, oculomotores, de la deglución y respiratorios, con inicio desde el nacimiento hasta la infancia temprana y una respuesta electromiográfica con decremento del potencial de acción muscular compuesto mayor al 10%.
Una vez establecida la sospecha, es importante buscar el diagnóstico específico a través de paneles genéticos, pues esto permite no solo la administración del tratamiento más adecuado, sino también establecer el pronóstico y la adecuada consejería genética a la familia.
Notas
Roles de autoría y contribución
IOE: conceptualización, escritura del artículo en español e inglés, revisión crítica de sus aspectos intelectuales y edición. Supervisión general y aprobación final de la versión completa en español. CR: conceptualización, escritura del artículo en inglés, revisión crítica de sus aspectos intelectuales y edición. GCh: revisión de aspectos genéticos, escritura del artículo y edición. AGE: conceptualización, escritura del artículo, revisión crítica de sus aspectos intelectuales y edición. Supervisión general y aprobación final de la versión completa en inglés.
Consentimiento informado
El consentimiento informado solicitado por Medwave para la publicación de este reporte de caso incluyendo las imágenes mostradas ha sido firmado por el padre del paciente. Los autores declaran que se respetó la privacidad del paciente según las normas de CIOMS, de privacidad de los datos recolectados. Una copia del consentimiento informado fue remitida a la dirección editorial de la revista.
Declaración de conflictos de interés
Los autores han completado el formulario de declaración de conflictos de intereses del ICMJE, y declaran no tener ningún conflicto de interés. Los formularios pueden ser solicitados contactando al autor responsable o a la dirección editorial de la Revista.
Financiamiento
El trabajo en el laboratorio del Dr. Engel fue financiado por el NIH NS6277 del National Institutes of Health de los Estados Unidos de America. El resto de los autores declaran que no se contó con fuentes de financiamiento externas para la elaboración de este reporte.
