Reporte de caso
← vista completaPublicado el 27 de febrero de 2020 | http://doi.org/10.5867/medwave.2020.01.7826
Reporte de un caso de síndrome de Noonan diagnosticado en atención primaria
A case report of Noonan syndrome diagnosed in primary healthcare
Resumen
El síndrome de Noonan es un trastorno genético de herencia autosómica dominante, de expresión fenotípica variable. Pertenece al grupo de las enfermedades conocidas como rasopatías, trastornos producido por las mutaciones en los genes RAS. Los pacientes desarrollan síntomas como dismorfismo facial, talla baja, enfermedad cardíaca congénita, alteraciones músculos esqueléticas y discapacidad intelectual. En el presente reporte, se describe un caso de diagnóstico del síndrome de Noonan en un paciente de 14 años, realizado a nivel de atención primaria en Ecuador. El síndrome se identificó mediante diagnóstico clínico, permitiendo su derivación al segundo y tercer nivel de salud para una atención especializada.
Ideas clave
|
Introducción
El síndrome de Noonan es un trastorno genético de herencia autosómica dominante de expresión variable, descrito por primera vez por la doctora Jacqueline Noonan en el año 1968 en una serie de 19 pacientes con las características propias del síndrome[1]. Esta patología pertenece al grupo de las enfermedades conocidas como rasopatías, trastornos producto de mutaciones en los genes que expresan para las proteínas RAS[2]. Estas intervienen en la ruta mediada por las proteínas quinasas activadas por mitógenos (MAPK)[3], controlando la proliferación, diferenciación, supervivencia y muerte celular[4],[5]. Clásicamente, el síndrome de Noonan se genera debido a alteraciones en los genes PTPN11, SOS1, RAF1 y KRAS[4], siendo las mutaciones del gen PTPN11 las más frecuentes, dando origen al 50% de los casos[6]. La identificación de las diversas alteraciones genéticas se realiza a través de métodos moleculares; los cuales poseen una sensibilidad del 70%3,[7].
El síndrome de Noonan afecta a uno de cada 1000 a 2500 recién nacidos vivos, presentando características similares al síndrome de Turner; por lo que también es denominado Turner con cromosomas normales[8]. Adicionalmente, presenta características típicas de la enfermedad, siendo estas dismorfismo facial, talla baja, enfermedad cardíaca congénita, alteraciones músculo esqueléticas y discapacidad intelectual[9],[10]. Las mismas que permiten establecer su diagnóstico sobre la base de los criterios clínicos propuestos por Van der Burgt (Tabla 1), para el diagnóstico clínico del síndrome de Noonan[11].
 Tamaño completo
Tamaño completo Considerando lo anterior, hasta 30% de casos positivos pueden no ser identificados mediante técnicas moleculares. Es por esta razón que el diagnóstico clínico continúa siendo una herramienta de gran relevancia a la hora de la determinación del síndrome de Noonan[7],[12]. La afección de dicho síndrome se ve reflejada en varios sistemas del cuerpo, por lo que es de vital importancia su diagnóstico oportuno y acertado para entregar un tratamiento integral y multidisciplinario[13]. En un centro de salud de atención primaria en Ecuador se identificó un paciente de 14 años que acudió por primera vez a la consulta de atención. En dicho establecimiento fue diagnosticado de este síndrome, básicamente debido a la clínica que manifestó.
Se presenta el caso de un paciente masculino de 14 años, nacido y residente en Ecuador, soltero, estudiante. La madre presentó amenaza de aborto a las 16 semanas de gestación y se acompañó de diabetes gestacional. El motivo de consulta fue dificultad para respirar más dolor en el pecho. El paciente acudió a consulta en compañía de su madre, quien refirió que su hijo presentaba desde hace más o menos un año como fecha real y cinco meses como fecha aparente, sensación de falta de aire durante la actividad física (clase funcional NYHA II), la misma que se ha incrementado progresivamente y cede con el reposo. Además, refiere que se acompaña de dolor torácico de intensidad leve a moderada, sin irradiación, el mismo que se ha exacerbado en los últimos días por lo que le fueron administrados analgésicos no especificados. Con ello, el cuadro presentó mejoría parcial razón por la que acude a consulta. Dentro de la revisión de aparatos y sistemas, el paciente presentó en los últimos cinco meses labilidad emocional.
El paciente se presentó consciente al examen físico; orientado en tiempo, espacio y persona; afebril.
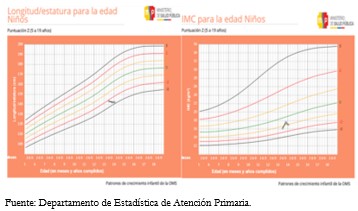
La valoración antropométrica fue peso: 36,3 kilogramos; talla: 1,45 metros, índice de masa corporal: 17,2 kilogramos por metro cuadrado. Así, el patrón de crecimiento que registró fue peso/edad bajo percentil 10, talla/edad -2 desviaciones estándar, e índice de masa corporal /edad -1 desviación estándar (Figura 1).
La cabeza presentó la dismorfia facial característica con presencia de hipertelorismo, cabello de implantación baja, pabellón auricular rotado y de implantación baja, puente nasal ancho. Los ojos con pliegues epicánticos, estrabismo (Figura 2A). Paciente presentó maloclusión dentaria (Figura 2B), cuello corto y ancho (Figura 2C). El tórax presentó deformidad en esternón tipo pectus excavatum (Figura 3).
En la auscultación de pulmones presentó murmullo vesicular conservado en ambos campus pulmonares. En corazón se auscultó soplo sistólico grado 2 a 3/6 en foco pulmonar ubicado en segundo espacio intercostal izquierdo; el segundo ruido cardíaco se expresó desdoblado amplio y fijo. Las extremidades eran simétricas, sin edemas, con amplitud de movimientos conservados. Sin embargo, se encontró el tono muscular disminuido e hipotrófico.
 Tamaño completo
Tamaño completo 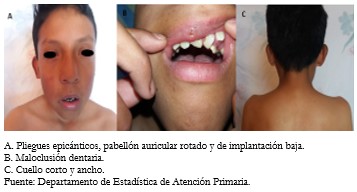 Tamaño completo
Tamaño completo 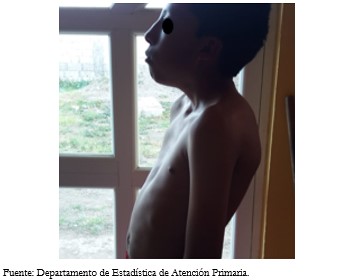 Tamaño completo
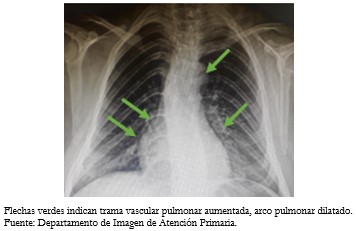
Tamaño completo Los exámenes realizados en atención primaria que se encontraban dentro de parámetros normales fueron biometría hemática, perfil renal, hepático y lipídico. La radiografía de tórax presentó ligera cardiomegalia a expensas de cámaras cardíacas derechas, con incremento de la trama vascular pulmonar y arco pulmonar dilatado (Figura 4).
 Tamaño completo
Tamaño completo El electrocardiograma reportó desviación del eje a la derecha, ritmo sinusal y hemibloqueo de rama derecha. El ecocardiograma reportó dilatación discreta de cámaras cardiacas derechas, defecto del septo inter-atrial tipo ostium secundum de 0,86 centímetros de diámetro y defecto interventricular subaórtico de 0,52 centímetros de diámetro. Presión sistólica de arteria pulmonar calculada en 46 milímetros de mercurio. Diagnóstico comunicación interauricular, comunicación interventricular, hipertensión pulmonar moderada (Figura 5).
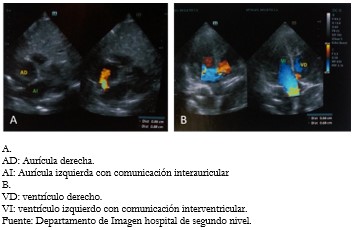 Tamaño completo
Tamaño completo El estudio psicológico reportó discapacidad intelectual leve y trastorno depresivo crónico. El examen de cariotipo fue normal. Luego del diagnóstico clínico establecido de síndrome de Noonan y los resultados obtenidos en el centro de salud de atención primaria, se realizó la referencia al hospital de segundo nivel para su valoración en las diferentes especialidades requeridas. Adicionalmente, se realizaron exámenes complementarios de especialidad y referencia a hospital de tercer nivel para asesoramiento genético e intervención quirúrgica de patología cardiaca. Al momento de redacción de este trabajo, el paciente se encuentra en tratamiento multidisciplinario en los tres niveles de atención.
Discusión
El síndrome de Noonan es considerado el trastorno más común entre las rasopatías, con un caso entre cada 1000 a 2500 recién nacidos[8]. En Ecuador se desconoce la prevalencia de dicha enfermedad, a pesar de que existen reportes en los años 2001 y 2011[14],[15]; sin considerar el descrito en el presente estudio. Esto puede deberse a que ciertos síntomas característicos, como las anomalías faciales, se vuelven menos evidentes conforme el individuo se acerca a la adultez[5]; por lo que se dificulta su identificación. Sin embargo, se puedo diagnosticar y derivar al paciente al segundo y tercer nivel de atención con el fin de obtener un tratamiento especializado.
El paciente presentó ciertas características típicas del síndrome de Noonan entre las que resaltan cara típica, pectus excavatum, cardiopatía congénita y talla por debajo de percentil 10. Así se cumplió con dos criterios mayores y dos criterios menores de los descritos por Van der Burgt, con lo que se estableció el diagnóstico clínico de síndrome de Noonan en el paciente. De igual manera, se reconocen características únicas de dicho síndrome como la dismorfia facial, cardiopatías congénitas, deformidades torácicas y estatura baja[16]. Estos criterios han permitido la exclusión del resto de rasopatías como el síndrome de Leopard, neuro-cardio-facio-cutáneo, Costello y neurofibromatosis-Noonan.
Asimismo, se pudo evidenciar características añadidas, como dificultad en el aprendizaje y alteraciones oculares, estos hallazgos se describieron en el estudio realizado por Altmüller y colaboradores[1], quienes evaluaron la presentación clínica en 17 pacientes con rasopatías de los cuales 15 presentaron un diagnóstico clínico de síndrome de Noonan. Dentro de los hallazgos clínicos se encontró que 94% de pacientes presentó dismorfia facial típica, 59% defectos cardiacos, 27% estatura baja y 42% discapacidad intelectual leve[1].
El síndrome de Noonan se caracteriza por tener afectación multisistémica, es así como en el estudio realizado por Şıklar y colaboradores[17] a 124 pacientes con síndrome de Noonan se encontró que 88,7% de ellos presentaron talla baja y características faciales típicas; 62,8% defectos cardíacos y 13,7% pectus excavatum. Así también se encontró discapacidad intelectual leve, hallazgos que también fueron encontrados en nuestro caso, siendo los rasgos faciales y la talla baja la afectación más frecuente en estos pacientes. Las personas que padecen de este síndrome durante el nacimiento presentan peso y longitud normales, mientras que durante la niñez y la adolescencia se evidencia talla baja. Se desconoce la causa de la presencia de este signo en el síndrome de Noonan[18].
Diversos estudios realizados demuestran que los rasgos dismórficos, talla baja, alteraciones esqueléticas, cardiopatía congénita, déficit cognitivo se presentan en todos los individuos con síndrome de Noonan[19]. Las cardiopatías congénitas son alteraciones en la estructura y funcionamiento del corazón, las mismas que están presentes desde el momento del nacimiento[20]. Del tres al 18% de los casos de cardiopatías congénitas están asociadas a síndromes genéticos; entre los más frecuentes se encuentran el síndrome de Down, síndrome de CHARGE (del inglés Coloboma, Heart defect, Atresia choanae, Retarded growth and development, Genital hypoplasia, Ear anomalies/deafness) y síndrome de Noonan[21]. El 80% de los casos de síndrome de Noonan presentan una patología cardiovascular, siendo esta la segunda particularidad más común[22].
Dentro de las afecciones cardíacas más comunes producidas por dicho síndrome son estenosis pulmonar presente en 75% de los pacientes y la miocardiopatía hipertrófica en 47% de los casos.
El paciente analizado no presenta las afecciones cardíacas más frecuentes. Sin embargo, tiene comunicación interauricular e interventricular, patologías que en la literatura se describe con una prevalencia de 32 y 16% respectivamente en personas con síndromes genéticos. No obstante, esta patología desencadenó síntomas que hicieron que el paciente fuera evaluado y diagnosticado de este síndrome en atención primaria[23].
De igual manera, se ha descrito una alteración poco frecuente en el síndrome, como es la maloclusión dental. La misma que puede traer complicaciones como dientes supernumerarios, mordida abierta, mordida cruzada posterior por lo que es importante su valoración odontológica oportuna[23].
Como se mencionó previamente, el diagnóstico de este caso se realizó tomando en cuenta los síntomas y signos clínicos del paciente. Se efectuó el análisis del cariotipo con el fin de esclarecer cualquier duda y de este modo excluir síntomas provocados por alteraciones cromosómicas, (Figura 4), donde se observó un patrón cromosómico normal. Así se confirmó que la enfermedad no se debe a variaciones cromosómicas.
Conclusiones
La atención primaria es la puerta de entrada al sistema de salud, de esta forma los pacientes diagnosticados oportunamente en el primer nivel de atención podrán ser remitidos para recibir valoración multidisciplinaria en el segundo y tercer nivel de atención en salud, de acuerdo con las patologías asociadas que presenten.
La adecuada evaluación clínica conduce a un tratamiento apropiado de la patología y un manejo integral de la misma, lo cual evita complicaciones posteriores que producirían un mayor gasto sanitario. Este es el caso de síndrome de Noonan, que tal como el presentado en este trabajo, ha demostrado que las características clínicas manifiestas en el paciente fueron fundamentales para su diagnóstico, basadas principalmente en la adecuada elaboración de la historia clínica. Adicionalmente, es importante reconocer que no existe un tratamiento específico para este síndrome, por lo que el tratamiento está enfocado en las patologías asociadas. Por esta razón, es indispensable su diagnóstico precoz, a fin de evitar complicaciones futuras.
Finalmente, se concluye que un adecuado manejo en el primer nivel de atención permitirá que el paciente reciba tratamiento quirúrgico de la patología cardiaca. Así, como también que se encuentra en proceso de calificación de discapacidad adecuada para la inserción escolar del paciente.
Notas
Contribuciones
Todas las autoras contribuyeron a la conceptualización, análisis formal, escritura y edición del manuscrito.
Agradecimientos
Las autoras expresan su agradecimiento a la Dirección de Investigación y Desarrollo y al Posgrado de Medicina Familiar y Comunitaria de la Universidad Técnica de Ambato.
Conflictos de intereses
Las autoras declaran no tener ningún conflicto de interés con los datos publicados.
Financiamiento
Este trabajo ha sido autofinanciado por las autoras.
Consentimiento informado
Este artículo cuenta con la autorización del tutor legal del paciente para su publicación.
