Publicado el 1 de enero de 2009 | http://doi.org/10.5867/medwave.2009.01.3695
Regulación del peso corporal
Regulation of body weight
Resumen
Este texto completo es una transcripción editada y revisada de una conferencia que se dictó en el V Congreso de la Asociación Chilena de Nutrición Clínica, Obesidad y Metabolismo realizado en Viña del Mar entre el 23 y 26 de Abril de 2008. El congreso fue organizado por la Asociación Chilena de Nutrición Clínica y Metabolismo, bajo la presidencia del Dr. Fernando Carrasco Naranjo.
Introducción
El balance de energía del organismo humano depende de la entrada y la salida de calorías; el exceso de ingesta que no sea utilizado en forma de energía se deposita en forma de grasa, lo que con el tiempo conduce a obesidad. La ingesta está regulada a nivel del hipotálamo por los centros del hambre y la saciedad, que a su vez tienen una serie de controles tanto metabólicos como no metabólicos (Fig. 1).
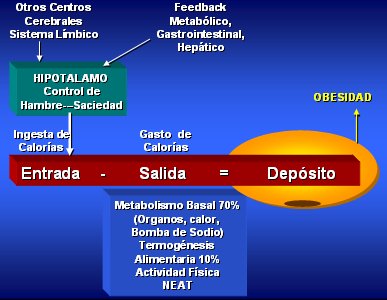 Tamaño completo
Tamaño completo El gasto energético en reposo se conoce como Metabolismo Basal; este gasto energético aumenta durante la ingesta, debido al efecto térmico de los alimentos, y durante la actividad física, sea como ejercicio propiamente tal o como consecuencia de la termogénesis de actividad involuntaria (NEAT = Non Exercise Activity Termogénesis). Este NEAT comprende los movimientos automáticos no voluntarios que ocurren durante las actividades cotidianas, el tono muscular, la mantención de la postura y los movimientos nerviosos involuntarios (fidgeting). Se ha observado que las personas obesas tienen menos movimientos automáticos involuntarios (1).
El apetito es un instinto muy complejo, necesario y placentero, pero muy difícil de controlar mediante la lógica o la voluntad. Los centros del hambre y la saciedad se ubican en el hipotálamo, pero éste tiene mútiples conexiones que influyen en el control del apetito a través de: las emociones como ansiedad, depresión, aburrimiento, frustraciones; el automatismo; factores culturales, familiares, costumbres; las características organolépticas de la comida como color, aroma, sabor, momento, lugar, horario, relación con fiestas, y la función cognitiva, que debería permitir el control voluntario, pero por desgracia no es así.
La actividad de comer tiene varios aspectos: el primero es el hambre, que cumple una función fisiológica cuando los depósitos de energía están muy bajos, conexiones del hipotálamo estimulan al centro del apetito y esto hace que aumente la ingesta y se mantenga el peso corporal. Luego está el apetito, que es el deseo de comer en función hedónica relacionado con características organolépticas de los alimentos. Después está el comer-no-motivado-por-hambre, que es un factor muy importante en la génesis de la obesidad. La gran mayoría de personas obesas no comen por hambre, sino por otros factores internos que aumentan la ingesta. Uno de estos factores es el comer automático, que consiste en comer sin darse cuenta y es la base del hecho de “si está enfrente, lo comemos”. Por este fenómeno tan peculiar es que todas las personas obesas, cuando se les pregunta cuánto comen, refieren cantidades de ingesta mucho menores que lo real; pero no mienten a propósito, sino que no se dan cuenta de la magnitud de su ingesta por el automatismo del comer.
Otro aspecto es el apetito emociogénico, que es en realidad un “espejismo” o “cortocircuito” cerebral, donde alteraciones emocionales tratan de conseguir una solución o reparación en la comida. Para la mente de muchas personas la comida no es solamente comida, sino que además tiene un valor simbólico muy importante, que conduce a esta gratificación oral. Estas personas sienten que comer es un placer y si la vida no les da algo o se los quita se consuelan con la comida, y lo que es peor, muchas veces no se dan cuenta de esto.
El apetito emociogénico lleva a la obesidad y la obesidad a su vez lleva a una mala adaptación psico-socio-económica que ocasiona baja autoestima, frustraciones, depresión y ansiedad. Una de las discriminaciones más difundida en todo el planeta es contra la obesidad, de modo que ahí se cierra el círculo vicioso, la mala adaptación social que aumenta el apetito emociogénico; como consecuencia el individuo come en exceso, se hace obeso y esto empeora su adaptación al medio ambiente. Comer fue la primera tentación y el pecado original; es más, Adán no comió la manzana porque tenía hambre.
En las reuniones de Overeaters Anonymous la primera pregunta que se hace es: “¿Ud. come cuando no tiene hambre?” Otro aspecto del comer que es muy importante es el “hábito alimenticio”, o sea, la manera de comer de todos los días. La gran mayoría de los seres humanos come el mismo tipo de comida y en la misma cantidad día a día. ¡Y es muy difícil cambiar los hábitos! La gratificación oral continuada conduce a la obesidad, la mala aceptación social del obeso aumenta su estrés y éste se transforma en ansiedad, depresión, aburrimiento, etc., sentimientos que por el cortocircuito del valor simbólico, no nutritivo, del comer cierran el círculo con más gratificación oral día a día. La industria alimentaria contribuye al aumento de la ingesta de calorías con el efecto subliminal de la publicidad, que asocia ciertos productos a felicidad, juventud, aceptación social y sonrisas, lo que favorece aún más el fenómeno equivocado de la gratificación oral.
La obesidad y la mentira
No existe área de la Medicina tan llena de mentiras como la obesidad. Entre la gran cantidad de mitos que se teje alrededor de este tema están: que la obesidad es una debilidad de la personalidad y que los obesos son glotones y culpables; que la obesidad sólo ocurre en los Estados Unidos; que la obesidad es un problema de comer mucho y que se puede tratar fácilmente; que la obesidad se puede prevenir después de una cena hipercalórica si se toma el café con aspartame; que la obesidad se cura con dietas muy eficaces o con un bypass gástrico; que la obesidad está siendo erradicada del planeta; y que se conocen perfectamente las causas y consecuencias de la obesidad. Los medios de comunicación contribuyen con mentiras más organizadas, bajo la forma de “tratamientos efectivos” con dietas, “medicamentos naturales” y aparatos de gimnasia.
Otro aspecto plagado de mitos es el uso de edulcorantes, cuyo impacto sobre la obesidad ha sido mínimo a pesar de que su consumo se ha promovido en forma masiva. Incluso algunos estudios sugieren que el consumo de edulcorantes aumenta el apetito debido al “Síndrome de la absolución metabólica imaginaria”, en que el individuo se convence a sí mismo de que al consumir estos productos está haciendo dieta, a pesar de seguir con una alta ingesta calórica. Otro auto-engaño muy común es el síndrome de la ingesta olvidada, en que los obesos no le asignan valor calórico a lo que comen, sino solamente valor emocional y se mienten a sí mismos mediante un mecanismo cerebral, no bien explicado pero muy común. La gran mayoría de los obesos están totalmente convencidos que comen una pequeña parte de todas las calorías que ingieren.
En resumen, pese a todo lo que se ha hecho para combatirla y a los múltiples tratamientos que se han propuesto, la obesidad sigue aumentando. Elliot P. Joslin dijo hace algunos años que el ser humano tiene demasiada grasa en su dieta, en su cuerpo y en sus arterias; hoy se puede añadir que le falta fibra en su dieta, en sus músculos y en su personalidad.
La masa adiposa del ser humano
La masa adiposa, es decir, la cantidad de grasa del organismo, depende del número y volumen de los adipocitos. Es importante destacar que muchas de las funciones de la grasa dependen del tamaño de los adipocitos; adipocitos pequeños y magros se comportan funcionalmente en forma muy diferente que adipocitos repletos y distendidos. La capacidad o capacitancia adipocítica, que es la máxima cantidad de triglicéridos que se puede acumular dentro de los adipocitos, depende del número y capacidad de distensión de cada uno de ellos. La capacitancia adipocítica es variable en distintos individuos, razas y edades y se cree que está determinada por factores genéticos, nutrición intrauterina, peso al nacer, etc. Las personas que nacen con bajo peso para la edad gestacional tienden a tener menor capacitancia adipocítica, al igual que los asiáticos, que con IMC de apenas 23 ó 24 comienzan a hacer traslocación de ácidos grasos libres (AGL) a músculo e hígado y desarrollan diabetes mellitus tipo 2.
La obesidad es una intoxicación crónica con calorías que se acumulan en forma de triglicéridos, inicialmente en los adipocitos, que por este motivo aumentan de tamaño, es decir, se distienden. El número de adipocitos queda fijo después de la pubertad; después de esa etapa vital la única variable que permite acumular más lípidos en el organismo es el aumento de volumen de estas células, lo que se traduce en cambios en su función endocrina. La obesidad es inicialmente una trigliceridosis adipocítica, o sea, una acumulación de lípidos dentro de los adipocitos, hasta que llega a sobrepasar la capacitancia adipocítica con la distensión máxima de cada una de estas células. Los adipocitos distendidos se hacen resistentes a la insulina, comienzan a hidrolizar los triglicéridos y liberan los AGL (“desborde”), que se traslocan hacia músculo, hígado y probablemente otros órganos.
No se ha determinado exactamente cuál es señal que media en este proceso; se ha planteado que puede radicar en el esqueleto de actina de la célula, pero el hecho es que cuando el adipocito alcanza su máxima distensión comienza a ser resistente a la insulina, aumenta la lipólisis, libera AGL y al mismo tiempo entra rápidamente en apoptosis, desencadenando una inflamación local en el tejido adiposo (adipositis). El aumento de tamaño del adipocito aumenta la traslocación de AGL hacia otros tejidos no adiposos y, al mismo tiempo, genera inflamación en el tejido graso. Los ácidos grasos que salen de los adipocitos penetran directamente las membranas de todas las células, sin necesidad de una orden hormonal. La glucosa, por el contrario, precisa de insulina, receptor insulínico, cascada fosforilativa y tráfico de los transportadores Glut-4, que se anclan en la membrana para poder entrar en la célula, de manera que su entrada está totalmente controlada por la insulina. Por el otro lado los AGL traspasan fácilmente la membrana celular debido a la estructura lipídica de ésta, de manera que cuando salen del adipocito ingresan directamente a músculo esquelético, donde el aumento del contenido de triglicéridos se asocia a una disminución de la captación de glucosa, e ingresan al hígado, aumentando la producción hepática de glucosa y de VLDL, característica de la obesidad y el síndrome metabólico.
Como ya se ha dicho, el aumento del tamaño del adipocito cambia su función endocrina. El tejido adiposo es el órgano endrocrino más grande del organismo y su función varía de acuerdo con el tamaño de los adipocitos. Adipocitos distendidos disminuyen la secreción de adiponectina, una hormona adiposa muy importante que disminuye la resistencia insulínica; si el tamaño del adipocito disminuye la adiponectina aumenta, lo que es beneficioso ya que disminuye la resistencia insulínica.
El tejido adiposo consta de diferentes tipos celulares: adipocitos y pre-adipocitos; fibroblastos y colágeno de soporte; macrófagos residentes y reclutados; células del estroma, células madre y otras células cuya función todavía se desconoce. Asimismo, se describen dos tipos de tejido adiposo: grasa blanca y grasa parda. La grasa blanca incluye la grasa subcutánea (gluteofemoral, de la pared abdominal y otra áreas), intraabdominal o visceral y otras; es amarillenta, aumenta con la edad, es metabólicamente activa, actúa como depósito de energía y su lipólisis libera ácidos grasos libres (AGL) a la circulación. La grasa parda es de color marrón rojizo debido a que contiene mayor cantidad de mitocondrias; se ubica a nivel interescapular, en roedores, y en tórax superior y cuello, en humanos; constituye 5% del peso en infantes humanos y después disminuye, pero no desaparece totalmente en los adultos, como se pensaba antes. La grasa parda es metabólicamente mucho más activa y no libera AGL porque libera calor, representando una fuente de termogénesis.
La grasa blanca toma glucosa y AGL y frente a la señal de disminución de insulina libera a dichos AGL para alimentar al resto del organismo, constituyendo un eficiente depósito de energía. La grasa parda también toma glucosa y AGL, pero en vez de liberar ácidos grasos los quema a través del efecto de las Proteínas Desacopladoras UCP y, de esa manera se queman grasas y se genera calor sin liberar AGL. El músculo es el más “egoísta” de los órganos desde el punto de vista energético, porque toma glucosa y AGL y lo único que “libera” es la contracción muscular. Por el contrario, el hígado es el más noble de los órganos metabólicos, porque toma glucosa, toma AGL, libera glucosa cuando es necesario y libera AGL en forma de triglicéridos y VLDL, que también se pueden reutilizar como fuente energética (Fig. 2).
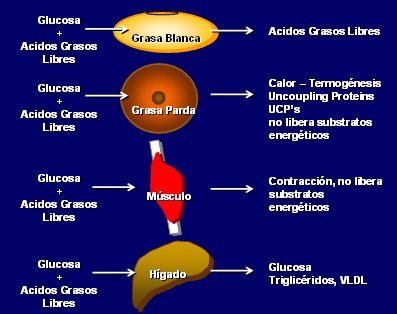 Tamaño completo
Tamaño completo Antes se pensaba que el adulto no tenía grasa parda, pero gracias a la moderna tecnología del PET scanning con FDG se ha demostrado que este tejido está presente en hasta 8 % de los adultos a nivel supraclavicular, cervical, mediastinal, suprarrenal, para-aórtica y paravertebral (no interescapular como los roedores). La lipólisis de la grasa parda es inducida rápidamente en un ambiente frío, por estimulación adrenérgica que se puede neutralizar con beta-bloqueantes (2, 3). Últimamente se ha encontrado que hay una proporción de grasa parda en el músculo. El rol de la grasa parda en el desarrollo de obesidad es motivo de intenso estudio actualmente.
Los adipocitos subcutáneos se acumulan más a nivel gluteofemoral, dando al cuerpo una forma de pera; aumentan por influencia de los estrógenos y el embarazo y permanecen por el resto de la vida; disminuyen por efecto de los andrógenos, por eso los hombres hipogonadales tienden a tener una imagen ginecoide. La grasa subcutánea también disminuye con los glucocorticoides, lo que explica la obesidad centrípeta del síndrome de Cushing, y secretan más leptina que los intrabdominales. La liposucción extrae adipocitos subcutáneos, pero esto no ocasiona baja de peso, porque las células que producen leptina disminuyen, los niveles de esta hormona bajan, aumenta el apetito y la persona vuelve a tener el peso anterior, con redistribución de la grasa.
El tamaño del adipocito regula sus funciones, que son: secreción de adipoquinas (adiponectina, resistina, etc). Con la distensión de los adipocitos aparece resistencia a la insulina y aumento de la lipólisis de los triglicéridos intra-adipocíticos. Esto conduce a la liberación de ácidos grasos y a su traslocación ectópica a músculo e hígado. Al mismo tiempo, la distensión extrema de los adipocitos hace que entren en apoptosis y ésto produce inflamación crónica del tejido adiposo, de bajo nivel, pero suficiente para incrementar tambien la lipólisis mediante el efecto local paracrínico de las citoquinas, especialmente el Factor Tumoral de Necrosis alfa, segregado por los macrófagos de la inflamación.
El adipocito distendido exporta AGL y resistencia insulínica; los AGL van directamente a otras células del organismo y: en el músculo disminuyen la captación de glucosa (fenómeno de Randle); en el hígado producen hígado graso, lo que aumenta la producción de glucosa y favorece la dislipidemia; y en la célula beta tendrían un efecto sobre su fatiga por lipotoxicidad. Por eso se dice que “¡La grasa debe permanecer en la grasa!”, porque cuando sale del adipocito al superar la capacitancia de éste se desarrollan los típicos problemas del síndrome metabólico (Fig. 3).
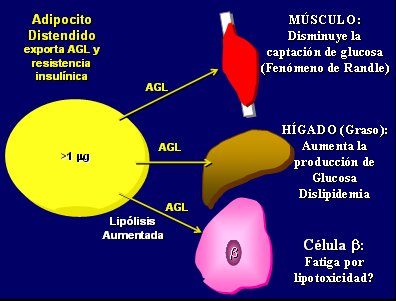 Tamaño completo
Tamaño completo No todas las funciones endocrinas del tejido adiposo se llevan a cabo en los adipocitos; los macrófagos y las células del estroma tienen también importantes funciones endocrinas. Por ejemplo, las citoquinas no son producidas por los adipocitos, sino por los macrófagos de la adipositis. Asimismo, la actividad de aromatasa de la grasa hace que el tejido adiposo se pueda considerar como un verdadero ovario periférico, ya que es el órgano más importante en la producción de estrógenos en la mujer post-menopáusica (4). La aromatasa, que convierte andrógenos en estrógenos, no está en los adipocitos, sino en las células del estroma (Fig. 4).
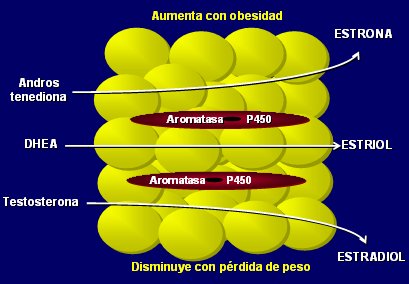 Tamaño completo
Tamaño completo Hoy, la nomenclatura ha cambiado y se define a las adipoquinas como hormonas del tejido adiposo, producidas por los adipocitos; y a las citoquinas, como sustancias activas producidas por las células inflamatorias del tejido adiposo (linfocitos, macrófagos) cuando se inflama por la obesidad. Los macrófagos del tejido adiposo se clasifican en dos tipos: los residentes, que están normalmente presentes en el tejido adiposo, y los reclutados, que son atraídos hacia el tejido adiposo desde la circulación por mecanismos poco conocidos, relacionados con el fenómeno de adipositis crónica propio de la obesidad (5). Ambos tipos de macrófagos se pueden identificar con marcadores celulares específicos.
Cuando el adipocito se agranda comienza a liberar sustancias que atraen a los macrófagos de la circulación y se inicia un fenómeno inflamatorio. Las personas delgadas tienen pocos macrófagos en su tejido adiposo y son sólo los macrófagos normalmente residentes, que no están activados y no segregan citoquinas. En los obesos la cantidad de macrófagos es mucho mayor, por el gran número de reclutados que van llegando desde la sangre (6, 7, 8). En el esquema de la siguiente figura, Hotamisligil muestra que cuando la persona aumenta de peso los adipocitos prácticamente “explotan”, provocando una reacción inflamatoria que atrae a los macrófagos a través de las moléculas MCP-1, que aumentan en el tejido adiposo hasta producir la inflamación crónica típica de la obesidad, al mismo tiempo que liberan citoquinas hacia la circulación sistémica, exportando la resistencia insulínica. Por lo tanto, el adipocito produce resistencia insulínica mediante dos mecanismos: la traslocación de AGL y la inflamación del tejido adiposo, ambos muy importantes (6) (Fig. 5).
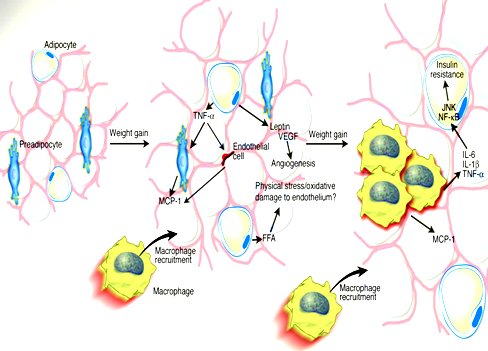 Tamaño completo
Tamaño completo Una de las formas más grave de resistencia a la insulina es la diabetes lipoatrófica, donde todos los triglicéridos están fuera de los adipocitos, de manera que no hay inflamación del tejido adiposo, sino sólo traslocación a otros órganos. A pesar de que no existe inflamación adiposa, se produce una resistencia insulínica muy grave.
La obesidad inflama a la grasa, produciendo una adipositis crónica; el reclutamiento de los macrófagos inicia la producción de varias citoquinas, lo que aumenta la producción de TNF alfa a nivel local y aumenta la resistencia insulínica a nivel sistémico. El TNF alfa tiene un efecto antiinsulínico importante. Al mismo tiempo, las citoquinas que se liberan a la circulación van hacia el hígado y aumentan la producción de PCR, fenómeno característico de las personas con obesidad, síndrome metabólico, resistencia insulínica y diabetes. En suma, la obesidad se asocia con inflamación crónica del tejido adiposo (adipositis) y aumento de la producción local de Monocyte Chemotactic Protein-1 (MCP-1), que va a atraer a los macrófagos solamente hacia el tejido adiposo, no hacia el hígado ni el músculo (9). Por otra parte, la obesidad aumenta el tamaño de estos adipocitos hasta que por un mecanismo desconocido “explotan”, en una apoptosis que termina en infiltración por macrófagos y adipositis (10).
El adipocito tiene una serie de receptores muy importantes que influyen en su función. Entre éstos está el receptor para el ácido nicotínico, cuya activación frena de inmediato la lipólisis en el adipocito, lo que explica su efecto hipolipemiante. En 2007 se descubrió que el ligando natural de este receptor es el cetoácido beta-hidroxibutírico, que aparece en la cetoacidosis y el ayuno prolongado, procedente del hígado, para actuar sobre el adipocito y disminuir la lipólisis, generando un sistema de retroalimentación perfecto entre lipólisis y disminución de la lipólisis cuando los niveles de beta-hidroxibutírico son muy altos (Fig. 6).
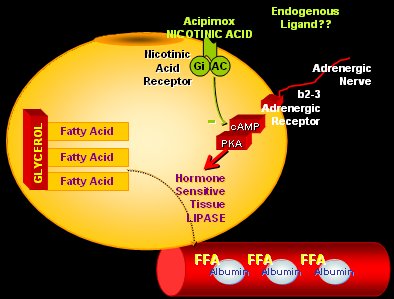 Tamaño completo
Tamaño completo De esta manera, en situación de ayuno o en diabetes tipo 1 descompensada, como ocurre en la cetoacidosis, disminuyen mucho los niveles de insulina, aumenta la estimulación adrenérgica, se estimula la lipasa hormono sensible en el adipocito y se inicia una lipólisis marcada. Los AGL que se liberan van rápidamente al hígado y estimulan a los receptores PPARalfa en el núcleo, aumentando el número y actividad de las mitocondrias y de los peroxisomas para que se realice la beta-oxidación de los AGL. De esta manera se pasa desde un combustible liposoluble a uno hidrosoluble, que es lo que se necesita en ese momento, por lo tanto, se cambia a los lípidos por los ácidos cetónicos, moléculas acuosas que van a actuar sobre este receptor de ácido nicotínico en la membrana del adipocito para disminuir la lipólisis. Así es como en una situación de ayuno prolongado se puede aumentar o disminuir la lipólisis en el tejido graso, de acuerdo a las necesidades. Este receptor también está presente en la piel y es por eso que cuando se administra ácido nicotínico se produce flushing y picazón (Fig. 7) (11, 12).
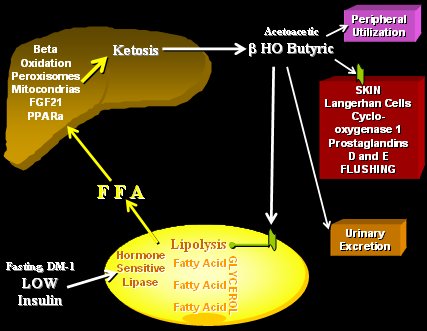 Tamaño completo
Tamaño completo Las funciones clásicas del tejido adiposo son: depósito de energía (sin agua); termogénesis (grasa parda); acolchado y relleno; aislamiento térmico; y depósito de vitamina D. Entre las funciones “nuevas” están: liberación de adipoquinas; regulación del metabolismo intermedio; regulación del eje gonadal; y actuar como “diana” de hormonas como estrógenos, andrógenos, hormona del crecimiento y glucocorticoides. Como depósito de energía es liposoluble y, por lo tanto, más liviano. Por otra parte, más que depósito de vitamina D el tejido adiposo es un “ladrón” de vitamina D, de modo que las personas obesas frecuentemente tienen una deficiencia de esta vitamina que debe ser suplementada. En todo paciente obeso se debe realizar determinación de 25-OH vitamina D en sangre, ya que 60 a 70% de ellos tienen niveles menores de 30 ng/dl y esto se agrava en los tiempos actuales, en que se ha disminuido la exposición solar para evitar las arrugas y el riesgo de cáncer. El déficit de vitamina D se agrava con la cirugía bariátrica, porque se produce una malaabsorción de esta vitamina liposoluble, de modo que en los pacientes que se han sometido a esta cirugía es indispensable aportar una dosis suprafisiológica de vitamina D, de 1000 a 10.000 unidades diarias ó 50.000 unidades por semana, para evitar el desarrollo de osteomalacia (13, 14, 15, 16).
Finalmente, es importante recordar que las modificaciones del estilo de vida pueden prevenir, curar o mejorar la obesidad y la diabetes.
