Congresos
← vista completaPublicado el 1 de septiembre de 2001 | http://doi.org/10.5867/medwave.2001.09.778
Estrógenos: mecanismos de acción de los estrógenos sobre el sistema cardiovascular
Mechanisms of action of estrogen on the cardiovascular system
Resumen
Este texto completo es la trancripción editada y revisada de la conferencia dictada en la XIV Reunión Científica de la Sociedad Interamericana de Hipertensión, 25-29 de marzo de 2001, Santiago.
Editora Científica: Dra. Gloria Valdés.
Introducción
En esta conferencia se estudiarán los distintos mecanismos de acción de los estrógenos sobre el sistema cardiovascular y, en la siguiente, se analizarán los resultados de algunos estudios clínicos sobre tratamiento de reemplazo hormonal en mujeres psmenopáusicas.
Existen muchas diferencias de sexo en las enfermedades vasculares y renales. Los hombres tienen la presión arterial más alta que las mujeres en los mismos grupos de edad. La enfermedad coronaria también es más frecuente en hombres que en mujeres premenopáusicas. Hay una mayor incidencia de enfermedades renales en hombres que en mujeres. Finalmente, el paso a insuficiencia renal es más rápido en hombres que en mujeres, con niveles similares de presión arterial.
Esta información da a entender que los hombres tienen una desventaja cardiovascular frente a las mujeres y que a éstas las protege el estrógeno.
Estradiol
Este estrógeno tiene funciones tanto a nivel del genoma (efectos genómicos) como en otros niveles (efectos no genómicos). Los primeros necesitan la síntesis de proteínas para la bioactividad del estrógeno; en cambio, los efectos que no son a nivel del genoma no necesitan la síntesis de proteínas para su bioactividad.
El estradiol se metaboliza, por lo menos, a tres metabolitos por acción del sistema del citocromo p450, de preferencia en los sistemas vascular y renal. El metabolito más dominante es el 2-hidroxiestradiol, que tiene alta afinidad con los receptores de estrógeno, es antioxidante y puede inhibir la catecol-o-metiltransferasa, con lo cual disminuye los niveles de catecolaminas. También es posible que tenga algún efecto anticarcinógeno.
El segundo metabolito del estradiol es el 4-hidroxiestradiol, que tiene muy baja afinidad con los receptores de estrógeno, pero su tasa de disociación del receptor es más lenta que la del 2-hidroxiestradiol. Según esto, los efectos biológicos del 4-hidroxiestradiol podrían ser mayores que los del 2-hidroxiestradiol. Es un co-oxidante, aumenta las prostaglandinas e induce la carcinogénesis.
El tercer metabolito es el 2-metoxiestradiol, que tiene acción antimitogénica en las células del músculo liso vascular, induce oxido nítrico (NO) y síntesis de prostaglandinas, inhibe la peroxidación de los lípidos y puede reducir el colesterol.
Efecto vasodilatador del estradiol
El estradiol produce un efecto vasodilatador agudo que se ejerce, en primer lugar, mediante el aumento de la actividad de la sintetasa de óxido nítrico (NOS). Este efecto no ocurre a nivel del genoma, por lo tanto no necesita un aumento en la síntesis de NOS; y se cree que estaría mediado por el receptor alfa de estrógeno. La mayor actividad de la NOS elevaría la producción de NO, llevando a un aumento de la guanilatociclasa y del GMP cíclico, que sería la causa directa de la vasodilatación.
Otro mecanismo por el cual el estradiol produce vasodilatación es la activación de distintos tipos de canales de potasio, como los canales de potasio activados por calcio, los canales BKCA, que son calcio y voltaje dependientes, y los canales Maxi-K, que están presentes en las células del músculo liso vascular.
También se ha demostrado que el GMPc, producido por el aumento del NO secundario a la mayor actividad de la NOS mediada por el estradiol, tendría un papel en el aumento de la actividad de estos canales de potasio y en la vasodilatación consecuente.
Un tercer mecanismo, que aumenta la vasodilatación en forma aguda, es el efecto del estradiol en el sistema de la adenilatociclasa. El estradiol acrecienta esta sustancia, la que produce un aumento del AMP cíclico, que a su vez eleva la adenosina, que causa la vasodilatación.
Efectos genómicos del estradiol
En cuanto a los efectos a nivel del genoma, se ha demostrado que el estrógeno aumenta la síntesis de NOS endotelial; esto es importante porque, además de causar vasodilatación, la NOS endotelial disminuye la agregación plaquetaria.
También se ha demostrado que el estradiol aumenta la síntesis de la NOS neuronal.
Al contrario de los efectos sobre la NOS endotelial y neuronal, el estradiol inhibe la producción de NOS inducible, cuya importancia radica en que produce concentraciones nanomolares de NO durante largos períodos, al contrario de las NOS constitutivas, que sólo producen concentraciones picomolares de NO durante lapsos breves.
Se ha visto que el NO producido por la NOS inducible en muchos sistemas inflamatorios se une con el superóxido; esta combinación de NO y superóxido tiene mayor afinidad que la combinación de superóxido con su degradador natural, la superóxido dismutasa. La velocidad de reacción de NO y superóxido es mucho mayor y, en términos termodinámicos, es más probable que la reacción entre superóxido y superóxido dismutasa. Entonces, el superóxido y el NO se combinan para producir un oxidante muy potente, llamado peroxinitrito, que tiene muchas funciones, entre ellas la producción de bFGF (Basic Fibroblast Growth Factor). El peroxinitrito también puede causar nitrificación de proteínas, lo que las inactiva, y puede causar quiebres en el DNA.
Con relación al tema de esta conferencia, una función importante del peroxinitrito consiste en inducir la producción del factor de crecimiento de fibroblastos, que es importante en la proliferación de células del músculo liso vascular. La inhibición de la síntesis de NOS inducible y el bloqueo de esta vía constituye uno de los principales mecanismos por los cuales este estrógeno protege contra la proliferación de las células del músculo liso vascular.
Siguiendo con los efectos del estradiol sobre la producción de NOS endotelial, hay evidencias clínicas de que la síntesis de NO disminuye en mujeres posmenopáusicas, lo que se ha demostrado analizando los metabolitos de nitrato y nitrito excretados en la orina de estas mujeres.
Por otra parte, el estradiol aumenta la vasodilatación coronaria mediada por acetilcolina. Esto se ha comprobado en primates, en mujeres posmenopáusicas en general, en mujeres posmenopáusicas que han tenido angina pero que no tienen enfermedad coronaria, y en grupos de transexuales de masculino a femenino.
Los estudios en transexuales se hicieron en Australia, utilizando una cohorte de mujeres y hombres transexuales. Estos reciben grandes cantidades de estrógenos para feminizarse, cuando son transexuales de masculino a femenino, y de testosterona para masculinizarse, cuando son de femenino a masculino. En ellos se observaron efectos cardiovasculares muy interesantes como resultado de la acción de las hormonas sexuales.
Se ha demostrado que el embarazo en ratas normales está asociado a un alto nivel de estrógeno, y por otra parte, la Dra. Bárbara Alexander ha informado que las concentraciones de NOS endotelial y proteica disminuyen en el riñón durante el embarazo, sin embargo, NOS neuronal y la NOS inducible aumentan durante toda la gestación. Por lo tanto, es importante que al discutir los efectos del estradiol se tome en consideración si es una situación fisiológica o patológica; se debe tener cuidado cuando se trata de utilizar in vivo información obtenida in vitro.
Inhibición de la mitogénesis de las células del músculo liso vascular
La inhibición de la mitogénesis por el estradiol se produce a través de dos mecanismos, uno de los cuales ocurre nivel del genoma y el otro, a otro nivel. Ambos son mediados por receptores de estrógenos.
En el mecanismo a nivel del genoma, el estradiol, mediado por los receptores de estrógeno, causa una disminución de la mitogénesis activada por proteína quinasa (MAPK), una enzima de fosforilación, reduciendo la transcripción de los factores c-myc y c-fos. La disminución de estos dos factores causa una reducción en la proliferación de las células del músculo liso vascular.
Además de la reducción de estos factores de transcripción, la disminución de c-myc y c-fos produce una disminución de IGF-1 y de los receptores de IGF-1, lo que lleva a una disminución de inositol fosfato, diacil-glicerol, proteína quinasa C y del calcio intracelular, todo lo cual también va producir una disminución de la proliferación de células de músculo liso vascular.
El mecanismo extra genoma del estradiol consiste en la activación de la adenilatociclasa, la que aumenta la actividad del AMP cíclico, que a su vez llevará a un aumento de la adenosina y por ende, a vasodilatación, y que también causará una disminución de la proliferación de las células del músculo liso vascular.
Otro mecanismo de acción importante del estradiol es el aumento del factor de crecimiento endotelial, así como también los receptores de este factor. El factor de crecimiento endotelial es importante en la endoteliogénesis, y el estradiol produce una recuperación rápida del endotelio en modelos de lesión mediante balón, e inhibe la formación de neoíntima.
Lamentablemente, debido al prominente efecto del estradiol sobre el factor de crecimiento endotelial, éste puede ser un mecanismo por el cual promueve ciertos cánceres de mama, ya que este factor es importante en la angiogénesis de ciertos tumores sólidos.
Se ha demostrado también que el estradiol bloquea la apoptosis de las células endoteliales inducida por especies oxígeno-reactivas y por TNFalfa. Esto es mediado por efectores de estrógeno, y por lo tanto sería un efecto genómico.
Efecto del estradiol sobre las moléculas de adhesión
Una vez que el endotelio es dañado, el resultado final es la proliferación de células de músculo liso vascular y la formación de placas. El estradiol, además de tener un efecto directo en la inhibición de esta proliferación, protege a las células endoteliales contra la lesión e inflamación, a través de una acción sobre la expresión de las moléculas de adhesión.
Generalmente, junto al daño endotelial se produce un aumento de la expresión de moléculas de adhesión y una activación subsecuente de células inflamatorias como leucocitos, macrófagos y monocitos. Una vez que estas células inflamatorias son activadas, se adhieren a las células endoteliales y activan la migración transendotelial, de modo que las células inflamatorias se localizan en el espacio subendotelial. Aquí los macrófagos fagocitan LDL y se transforman en células espumosas, las que liberan factores de crecimiento que favorecen la proliferación de células de músculo liso vascular y la formación de la placa (Figura 1).
 Tamaño completo
Tamaño completo Figura 1. Injuria endotelial y formación de la placa.
Por otra parte, se ha demostrado que el estradiol puede inhibir la expresión de las moléculas de adhesión E-selectina, ICAM-1 y VCAM, lo que constituye otro mecanismo de protección.
Efectos del estradiol en el sistema renina-angiotensina
Mediante un efecto genómico, el estradiol aumenta la producción hepática de angiotensinógeno, el que es convertido a angiotensina II (ATII) en el plasma por acción de la renina. Esto debería llevar a un aumento de la ATII, pero se ha visto que el estradiol produce una disminución de los receptores AT1 en la aorta. Un estudio reciente realizado por Nickenig demostró que la ooforectomía estimula el RNA mensajero para receptores de AT1, mientras que el tratamiento con estradiol reduce el número de dichos receptores (Circulation 1998;97:2197-201).
Además, el estradiol causa una disminución de la actividad de la enzima convertidora de angiotensina, lo que lleva a una disminución de la ATII (Figura 2). Sin embargo, la actividad de la enzima convertidora no sería el paso limitante para la conversión de angiotensina a angiotensina II, sino que sería más bien el efecto de renina.
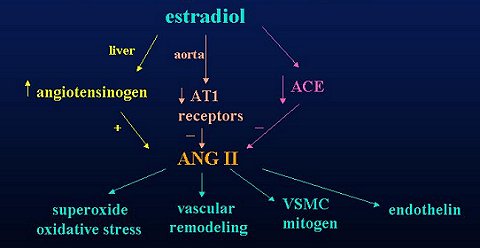 Tamaño completo
Tamaño completo Figura 2. Efectos del estradiol sobre el sistema renina-angiotensina.
Por otra parte, se ha demostrado que la ATII produce un aumento del superóxido y del estrés oxidativo, al aumentar algunas de las subunidades de NADPH oxidasa. También causa un aumento del factor de crecimiento tisular beta, del remodelamiento vascular y de la proliferación de las células del músculo liso vascular, además de un aumento de los niveles de endotelina.
Todos estos factores afectan al sistema vascular, y por lo tanto afectan a las enfermedades cardiovasculares y renales. Si el estradiol puede proteger contra los aumentos de angiotensina II, teóricamente podría proteger contra todos estos efectos en el sistema vascular.
Las mujeres posmenopáusicas tienen generalmente niveles de renina plasmáticos más altos que las mujeres premenopáusicas, y la actividad de renina es mayor en hombres que en mujeres posmenopáusicas. Sin embargo, las mujeres que reciben tratamiento hormonal tienen actividad de renina plasmática menor que mujeres de la misma edad sin tratamiento, según un estudio hecho por Schunkert (Circulation 1997;95:39-45).
Otro factor de riesgo vascular sobre el cual el estradiol podría actuar, es sobre los niveles de homocisteína circulante, que son reducidos por efecto del estrógeno. Esto se vio en mujeres menopáusicas y en transexuales de masculino a femenino, a diferencia de los andrógenos, que tienen efectos opuestos.
Con respecto a los lípidos, que es otro factor de riesgo importante, el estradiol reduce la oxidación de LDL y VLDL, aumenta los receptores de LDL, disminuye la producción de LDL y puede aumentar el HDL, principalmente a través de sus efectos sobre la apoproteína A1. Sin embargo, se ha visto que aumenta los niveles de triglicéridos.
Estudios clínicos
Con respecto al papel protector del estrógeno contra el aumento de la presión arterial, en un estudio realizado por Wiinberg se tomó la presión arterial en forma ambulatoria a una población danesa normotensa de 352 hombres y mujeres de distintas edades. Se vio que la presión arterial aumentaba con la edad y que era notoriamente mayor en los hombres en todos los grupos etarios hasta los 70-80 años de edad, donde la curvas se juntaban (Figura 3) (Am J Hypertens 1995 Oct;8(10 Pt 1):978-86).
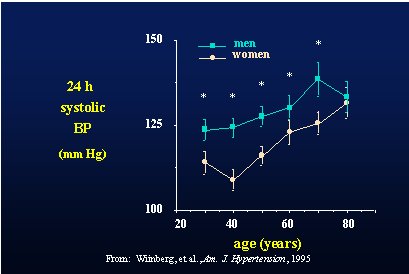 Tamaño completo
Tamaño completo Figura 3. Presión arterial sistólica según edad y sexo.
En un informe del National Health Nutrition Evaluation Survey de los Estados Unidos se describe la prevalencia de hipertensión en tres cohortes formadas por negros no hispanos, blancos no hispanos, y mejicano-americanos. En los negros no hispanos se vio que en las edades de 50 a 59 años, los hombres tenían un prevalencia más alta que las mujeres (la menopausia en promedio ocurría a los 51,4 años de edad), pero a los 60 a 69 años de edad ocurría un aumento significativo en las mujeres, que persistía a lo largo del envejecimiento.
En los mejicano-americanos, entre los 50 y 59 años de edad las mujeres ya tienen una prevalencia de hipertensión similar a los hombres, y hay un aumento en la prevalencia de hipertensión con la edad. El problema con estas dos cohortes es que la información está afectada por la alta incidencia de obesidad y diabetes mellitus tipo 2, que causan hipertensión.
En la población de blancos no hispanos, donde la incidencia de obesidad y diabetes mellitus tipo 2 no son tan altas, los hombres tienen una prevalencia más alta de hipertensión a los 50-59 años de edad, la prevalencia es similar a los 60-69 años de edad, y cuando las mujeres llegan a la edad de 70-79, tienen una prevalencia más alta de hipertensión que los hombres.
A raíz de estos hallazgos cabe preguntarse por qué, si la menopausia llega a los 51,4 años de edad, se demora alrededor de 10 años en aumentar la presión arterial. Esto sugiere que el déficit de estrógeno podría estar jugando un papel importante en el aumento de la presión arterial en mujeres posmenopáusicas.
Edición científica a cargo de la Dra. Gloria Valdés, Hospital Clínico Pontificia Universidad Católica de Chile.
