Cursos
← vista completaPublicado el 1 de diciembre de 2004 | http://doi.org/10.5867/medwave.2004.11.3367
Distrofias musculares de Duchenne y de Becker: aspectos genéticos
Duchenne and Becker muscular dystrophy: genetic aspects
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
Las distrofias musculares de Duchenne (DMD) y de Becker (DMB) son patologías neuromusculares degenerativas frecuentes, graves e intratables, que producen debilidad y deterioro progresivo de los músculos esqueléticos, respiratorios y cardíacos (1). Ambas enfermedades son ocasionadas por mutaciones del gen, localizado en el brazo corto del cromosoma X, que codifica para la proteína distrofina, polipéptido esencial para la mantención estructural y mecánica de la fibra muscular. Su gravedad y pronóstico asociado dependen principalmente de las características de las mutaciones en este gen (2).
En la presente revisión se analizarán los aspectos clínicos, las bases genéticas, la incidencia dentro de la población, el diagnóstico y el consejo genético de estas enfermedades, con especial énfasis en sus bases genéticas y hereditarias.
Descripción clínica
La DMD está presente desde el nacimiento y se manifiesta clínicamente muy temprano en la infancia del paciente, de manera que antes de los 5 años de edad ya es posible detectar la pérdida de la fuerza muscular, que es progresiva y afecta predominantemente a la musculatura proximal de los miembros y a los músculos flexores del cuello. La afección de las piernas es más evidente que la de los brazos.
Los individuos afectados presentan pseudohipertrofia de la musculatura de la pantorrilla y el clásico signo de Gower; hacia los 11 a 12 años de edad, la mayoría de ellos permanece en silla de ruedas, las contracturas se estabilizan y a menudo aparece una escoliosis progresiva, que ocasiona problemas pulmonares. Casi todos los pacientes desarrollan miocardiopatías y cierto grado de deficiencia cognitiva, con coeficientes intelectuales disminuidos en una desviación estándar con respecto al promedio de la población (2). La muerte ocurre por falla cardíaca o respiratoria, antes de los 30 años de edad.
Los síntomas y signos presentan menos gravedad en la DMB, cuya aparición ocurre en una edad más avanzada. Por definición, los pacientes que pueden caminar después de los 15 años de edad, presentan DMB. La capacidad para caminar puede continuar en la edad adulta, hasta los 40 años (1).
Bases genéticas
Ambas enfermedades tienen un patrón de herencia recesiva ligada al sexo, de modo que una mujer debe tener ambos alelos mutados para presentar la enfermedad; si tiene sólo un alelo mutado, será sólo portadora. En cambio, un hombre con un alelo mutado siempre será afectado; es por esto que la DMD y DMB afectan principalmente a los varones (2). Aproximadamente dos terceras partes de los individuos heredan la enfermedad de sus madres y el tercio restante se produce por la aparición de una nueva mutación. Respecto al concepto de penetrancia, ambas distrofias presentan penetrancia completa es decir, si se presenta el genotipo característico de la patología, siempre se expresará la enfermedad (3).
La expresividad de esta patología es muy variada, originando dos enfermedades distintas, dependiendo de la magnitud de los síntomas y signos que se presenten. Se habla de DMD cuando el cuadro es muy grave y se produce tempranamente y de DMB cuando es leve y se expresa más tardíamente.
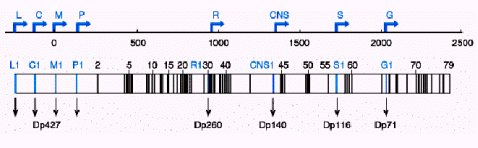
El gen de la distrofina, localizado en el cromosoma X, específicamente en Xp21, está formado por 2,4 millones de pares de bases, lo que lo convierte en el gen más grande que se ha encontrado en humanos, ya que representa el 1,5% del cromosoma X. El 0,5% del gen, o sea, 13.973 pares de bases, contiene los 79 exones que codifican para la síntesis de la proteína distrofina, de 427 kD (véase Figura 1) (2).
 Tamaño completo
Tamaño completo Figura 1. Esquema del gen de la distrofina, que codifica para las 8 isoformas de la proteína distrofina. Tiene un tamaño de 2,4 MB, se encuentra ubicado en el cromosoma X (Xp21), posee 79 exones y 9 promotores.
La transcripción del gen de la distrofina en ARNm está bajo el control de 8 promotores, que gobiernan el proceso de expresión en distintos tejidos, generando distintos tipos de proteínas. El producto principal del proceso de traducción de este gen es la distrofina de extensión completa, una proteína muy larga, con forma de varilla, constituida por 3.685 aminoácidos. La distrofina es parte de los costámeros, que conectan los discos Z del sarcómero, las unidades contráctiles, con el sarcolema o la membrana celular.
Por esto, la presencia de distrofina es esencial para la estabilidad de las células musculares durante la contracción muscular, la que da lugar a una deformación celular y al acortamiento de las miofibrillas. Durante este proceso, la maquinaria contráctil dentro de las miofibrillas debe seguir conectada íntimamente con la membrana y la matriz extracelular; sin esta asociación el movimiento sería transmitido incorrectamente y las células musculares podrían dañar sus membranas. La función del complejo de la glicoproteína Distrofina (DGC) es proporcionar un acoplamiento mecánico fuerte del citoesqueleto intracelular a la matriz extracelular.
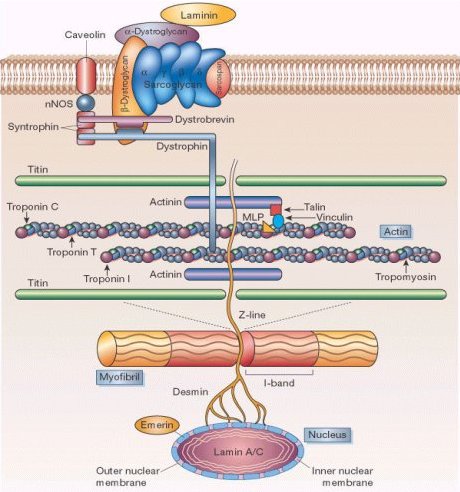
El DGC se compone de proteínas de transmembrana citoplásmicas y extracelulares, de las cuales se conocen más de 50, entre ellas, la distrofina, los sarcoglicanos, las sintrofinas, las integrinas, distrobrevina y la oxido nítrico sintetasa, entre otras (véase Figura 2). Cuando la distrofina está ausente, el balance entre las diferentes partes de este complejo distrófino (ligado a la distrofina) se perturba, afectando especialmente a los distroglicanos, los sarcoglicanos y sarcospan, que se reducen o desaparecen completamente.
 Tamaño completo
Tamaño completo Figura 2. Esquema del complejo Distrofina-Glicoproteínas. Se observa la proteína distrofina asociada en un extremo a la F-actina y en su otro extremo, al beta-distroglucano. De esta manera la proteína distrofina proporciona el acoplamiento mecánico entre el sarcolema y el citoesqueleto de la fibra muscular. (Tomado de http://www.abc-online.org/forsch/scheub01b.jpg).
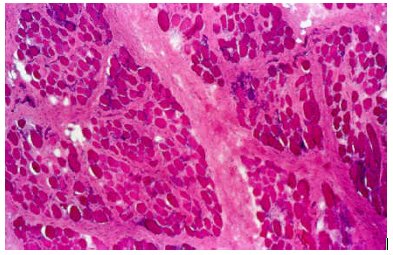
La DMD se produce debido a mutaciones en el gen de la distrofina. Como consecuencia de estas mutaciones se transcribe un ARNm con un marco de lectura alterado, lo que origina una proteína trunca (no funcional), que es rápidamente degradada, con la consiguiente ausencia de distrofina y alteración de todo el complejo Distrofina-Glucoproteínas. Estas alteraciones conducen a la pérdida de la unión entre el citoesqueleto y el sarcolema y, finalmente, a la necrosis de la fibra muscular (ver Figura 3).
 Tamaño completo
Tamaño completo Figura 3. Corte histológico de tejido muscular esquelético de un paciente con distrofia muscular de Duchenne (tinción de hematoxilina-eosina). Se observa variación en el tamaño de las fibras, necrosis y marcada proliferación de tejido conjuntivo. (Tomado de Enfermedades del músculo estriado, http://www.scn.es).
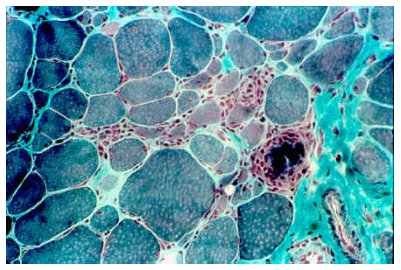
La DMB se produce debido a la ocurrencia de mutaciones que, en el 95% de los casos conocidos, no ocasionan alteraciones en el marco de lectura, de modo que permiten la producción de distrofina, la cual se encuentra alterada, pero no ausente. Esto debilita la unión entre el sarcolema y el citoesqueleto, por lo que la fibra disminuye de tamaño lenta y progresivamente, hasta producirse necrosis del tejido (4) (véase Figura 4). Las mutaciones que afectan al gen de la distrofina corresponden en un 67 % a mutaciones heredadas y en un 33% a mutaciones nuevas, y del total de las mutaciones, dos tercios corresponden a deleciones y el otro tercio, a duplicaciones y traslocaciones.
La heterogeneidad genética se define como la existencia de diferentes mutaciones en un solo locus (mutaciones alélicas), o en diferentes locus (mutaciones no alélicas), que producen un síndrome clínico similar; en la DMD y DMB se puede apreciar claramente este fenómeno, debido a que ambas presentan signos y síntomas similares, de gravedad diferente, debido a distintos tipos de mutaciones en el mismo gen.
 Tamaño completo
Tamaño completo Figura 4. Corte histológico de tejido muscular esquelético de un paciente con distrofia muscular de Becker. Se observa variación de tamaño de las fibras y fagocitosis (Tinción de tricrómico modificado de Gomori). (Tomado de Enfermedades del músculo estriado, http://www.scn.es).
Incidencia
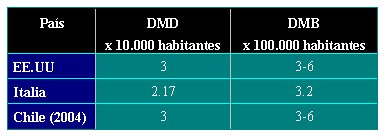
En términos geográficos, y a modo de ejemplo, en los Estados Unidos la incidencia de la DMD es de 3 por cada 10.000 habitantes y la de la DMB, de 3 a 6 casos por cada 100.000 personas. En Canadá, la frecuencia de DMD en la población es de 2,92 por 10.000 personas (5), mientras que en Italia la DMD presenta una incidencia de 2,17 por cada 10.000 personas y la DMB, de 3,2 por cada 100.000 habitantes (6).
No existen grandes diferencias en las tasas de DMD entre distintos países o grupos étnicos (7); en Chile, la incidencia de DMD y DBM es de 3 afectados por cada 10.000 habitantes y de 3 a 6 afectados por cada 100.000, respectivamente (véase Tabla 1).
 Tamaño completo
Tamaño completo Tabla 1: Comparación de la incidencia de DMD y DMB en Chile, Italia y Estados Unidos.
Diagnóstico y manejo
El diagnóstico se efectúa, fundamentalmente, en dos etapas. Inicialmente, cuando el médico tiene la sospecha de que la patología está presente, porque el paciente presenta problemas motores, se realiza un diagnóstico clínico y a través de exámenes de laboratorio. El primero de ellos es la cuantificación del nivel de creatinfosfokinasa sérica (CPK), observándose que los pacientes con DMD tienen valores 50 a 100 veces por sobre lo normal, debido al escape de esta proteína desde la célula por la ruptura de la fibra. Además se debe realizar biopsia muscular y electromiografía, examen que permite determinar el grado de conducción del nervio y descartar la presencia de una enfermedad neurodegenerativa.
La siguiente etapa consiste en corroborar el diagnóstico mediante un análisis genético, que consiste en la amplificación simultánea de diferentes regiones del gen de la distrofina mediante la técnica de PCR. A través de este análisis genético se puede realizar un diagnóstico prenatal, cuando existe la sospecha de que el hijo en gestación presenta la enfermedad.
Es importante saber que, en nuestro país, el 85% de los casos de DMD se diagnostica tardíamente, debido a que los padres consultan tardíamente y los médicos generales derivan tardíamente los casos al médico especialista (8).
El consejo genético consiste en explicar al paciente y/o a sus familiares cuál es la evolución del defecto de nacimiento o enfermedad que motiva la consulta, el manejo terapéutico o de rehabilitación del mismo y, sobre todo, informar sobre las medidas preventivas que se pueden tomar. Es necesario informar a los padres sobre el riesgo de recurrencia de la mutación en futuros hijos, para lo cual es necesario identificar a los portadores en la familia. Se debe realizar un diagnóstico a tiempo para tomar las medidas del caso en cuanto actividad física, terapias, desarrollo psicosocial, etc.
En cuanto a tratamiento, para las distrofias musculares de Duchenne y Becker no existe un tratamiento que permita curar la enfermedad. Actualmente se está investigando la aplicación de diversos tipos de terapia génica, pero por ahora no hay resultados concluyentes (9).
