Cursos
← vista completaPublicado el 1 de noviembre de 2004 | http://doi.org/10.5867/medwave.2004.10.3369
Enfermedad de Huntington: aspectos genéticos
Huntington's disease: genetic aspects
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
Descripción y características clínicas
La enfermedad de Huntington, o corea de Huntington, es una enfermedad neurológica degenerativa y hereditaria. Las principales características de la enfermedad son la presencia de movimientos involuntarios de tipo coreico y las alteraciones psiquiátricas, cognitivas y del comportamiento (Tabla I).
 Tamaño completo
Tamaño completo Tabla I: Principales síntomas de la enfermedad de Huntington (4).
Clásicamente se considera al corea, síndrome hipercinético caracterizado principalmente por movimientos involuntarios amplios, irregulares, generalizados y bruscos, como el cuadro motor más característico, aunque no es el único tipo de alteración del movimiento descrito. Las mayores limitaciones de los pacientes más evolucionados son la distonía, la rigidez y las alteraciones de la marcha, que aparecen en el curso evolutivo de la enfermedad. Los sujetos que inician los síntomas en edad infantil y juvenil suelen desarrollar una variante rígido-acinética de la enfermedad que se conoce como forma de Westphal (1).
También son características de la enfermedad las alteraciones de la motilidad ocular, sobre todo de los movimientos sacádicos (1). Asimismo, se puede presentar disartria (incapacidad para articular palabras apropiadamente), disfagia (dificultad para deglutir), inestabilidad postural, ataxia (debilidad, descoordinación y temblor intencional), mioclonía (contracción súbita, no rítmica, de uno o más músculos o parte de un músculo) y tics.
Las alteraciones psiquiátricas son muy variadas e incluyen cambios de personalidad, apatía, agitación, impulsividad, depresión, manía, paranoia, hostilidad, agresividad, alucinaciones y psicosis. Los trastornos cognitivos (demencia) se manifiestan fundamentalmente por pérdida de la memoria reciente, juicio pobre, alteración de la concentración y de adquisición de nuevos conocimientos.
En cuanto a los cambios en el comportamiento, los pacientes presentan retraimiento, apatía y tendencia al mutismo. Sin embargo, en un entorno convenientemente estructurado que les facilite estímulos adecuados, se consigue que muestren interés en las actividades diarias. También pueden aparecer otros síntomas como menor flexibilidad mental y menor agilidad psicomotora, con inhibición de las respuestas automáticas y alteración en la formación de conceptos. El paciente puede parecer menos atento, menos espontáneo y con embotamiento afectivo. Sin embargo, su capacidad emocional permanece intacta, por lo que hay que vigilar posibles intentos de suicidio, frecuentemente no exteriorizados.
La sintomatología clínica del paciente puede variar según la edad de comienzo de la enfermedad o el momento evolutivo del proceso. Clásicamente se ha considerado que las formas infantiles, que inciden sobre un cerebro inmaduro y en formación, presentan mayor grado de demencia, con mayor grado de lesión histológica del cerebro, mientras que en las formas seniles se suelen encontrar deterioros cognitivos leves; sin embargo, la mayoría de los estudios clásicos describen la severidad del deterioro según la alteración de las actividades de la vida diaria y por la percepción subjetiva de los familiares, factores que están muy interferidos por las alteraciones motoras presentes. En la Tabla II se resumen las diferencias entre las formas de comienzo en edad juvenil y tardía de la enfermedad (1).
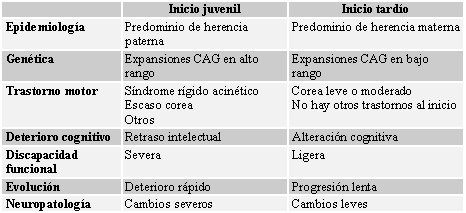 Tamaño completo
Tamaño completo Tabla II: Diferencias entre la forma de inicio de la enfermedad de Huntington en edades juvenil y tardía (1).
Bases genéticas
La enfermedad de Huntington se transmite de forma autosómica dominante, característica que ha posibilitado un mejor conocimiento de sus causas y, principalmente, de sus alteraciones moleculares. En 1983 se localizó la región del genoma involucrada en la enfermedad en el cromosoma 4, específicamente en la región cromosómica 4p16.3 (7); más adelante, en 1993, fue identificado el gen responsable de la enfermedad, el gen IT15, que se extiende en una región del ADN de aproximadamente 180 kb, contiene 67 exones y porta una amplificación génica producida por la repetición del trinucleótido CAG.
El gen codifica para un mRNA de 10 kb que se traduce como una proteína de 350 kDa, la huntingtina(5), que se encuentra en todas las neuronas del cerebro y cuya función normal es desconocida (6). La mutación genética que provoca la enfermedad consiste justamente en la amplificación del trinucleótido CAG, en un segmento del ADN que es inestable y está localizado en el exon 1 del gen. La expansión del trinucleótido repetido (CAG)n origina la enfermedad de Huntington por su efecto sobre la expresión o estructura de la proteína codificada por el gen 1T15.
El gen IT15, en los individuos normales, contiene entre 15 y 34 repeticiones de CAG, en cambio, el gen en los pacientes con enfermedad de Huntington contiene desde 35 hasta más de 66 repeticiones. Como el trinucleótido CAG codifica para glutamina, esto hace que se produzcan huntingtinas con una cadena anormalmente larga de glutaminas (7).
Cada hijo de un padre que presenta el trastorno tiene un 50% de posibilidad de heredar la enfermedad de Huntington. Los síntomas generalmente no aparecen hasta la edad adulta, normalmente entre los 35 y 50 años, sin embargo, esto depende del número de copias de CAG presentes en el gen, pudiendo aparecer el trastorno en personas más jóvenes si el número de copias de CAG es alto; de esta manera, el número de repeticiones del trinucleótido CAG predice el riesgo de desarrollar la enfermedad (Tabla III).
 Tamaño completo
Tamaño completo Tabla III: Riesgo de desarrollar EH según el número de repeticiones de CAG.
Del mismo modo, el número de copias de CAG puede determinar la gravedad de la enfermedad, ya que las personas con un bajo número de copias tienen movimientos anormales leves, mientras que las personas con un número elevado de copias están severamente afectadas (3).
Rubinsztein y cols. (1996) reportaron que seis individuos, aparentemente normales, poseían desde 36 hasta 39 repeticiones de CAG, demostrando así que el gen mutado no presenta penetrancia completa en todos los casos (8).
La expresividad del gen está dada por la intensidad en la manifestación de la enfermedad; así, una expresividad alta se producirá en aquellos individuos que presentan un alto número de repeticiones CAG, y una expresividad baja se producirá en aquellos pacientes que tienen un bajo número de repeticiones.
Patogénesis de la enfermedad
Específicamente, las células afectadas son las del ganglio basal, una estructura profunda del cerebro que tiene importantes funciones, incluyendo la coordinación de los movimientos. En el ganglio basal, la enfermedad de Huntington ataca específicamente a las neuronas del cuerpo estriado, especialmente a las del núcleo caudado y del globo pálido. También está afectada la corteza cerebral, que controla el pensamiento, la percepción y la memoria.
Los rasgos anatomopatológicos característicos son la atrofia severa bilateral de los núcleos caudado y del putamen, con atrofia moderada de los lóbulos frontales y temporales (véase figura 1). A nivel microscópico se ha demostrado pérdida selectiva y precoz de pequeñas interneuronas espinosas gabaérgicas en los núcleos caudado y en el putamen, y pérdida de neuronas grandes en el globo pálido. La corteza cerebral muestra grados variables de estrechamiento cortical, pérdida neuronal, distorsión de la arquitectura y gliosis reactiva.
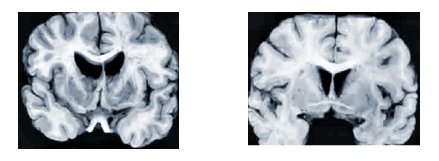 Tamaño completo
Tamaño completo Figura 1. Alteraciones macroscópicas producidas por la enfermedad de Huntington. En un individuo con EH (imagen izquierda) se aprecia atrofia del núcleo caudado y del putamen. La imagen derecha corresponde a un individuo control normal (11).
En las fases iniciales de la enfermedad se afectan las zonas anteromediales del cuerpo estriado, progresando la lesión hacia zonas posteriores y laterales de este cuerpo en fases más tardías de la enfermedad. En los pacientes que presentan inicio juvenil de la enfermedad los cambios histológicos son más severos y se extienden al tálamo, cerebelo e incluso a los tractos corticoespinales. La severidad de los cambios histológicos parece relacionarse directamente con la severidad del defecto molecular.
Las consecuencias bioquímicas de estas alteraciones neuropatológicas son la disminución del ácido gamma-aminobutírico (GABA) y de la actividad de la enzima descarboxilasa, responsable directa de la síntesis del ácido glutámico (GAD), en núcleo caudado y putamen y, en menor medida, en globo pálido, núcleo subtalámico, corteza y troncoencéfalo. También se ha descrito pérdida paralela de neuronas productoras de encefalina, que sería el sistema neurotransmisor que se compromete en forma más precoz, sustancia P y enzima conversora de angiotensina, y alteraciones en el metabolismo de la dopamina (1).
El origen molecular de la enfermedad se debería a que un exceso de repeticiones de CAG en el gen IT15 es capaz de cambiar la configuración de la proteína huntingtina producida por éste. Cada trinucleótido CAG codifica para un aminoácido glutamina; con un número mayor de glutaminas que lo normal, la huntingtina adquiere una configuración anormal. La huntingtina mutante interactúa con todo tipo de proteínas, la mayoría de las cuales, normalmente, son ignoradas; esto ocurre particularmente en neuronas espinosas de estructuras cerebrales profundas.
Se cree que alguna de estas proteínas es capaz de escindir a la huntingtina mutante en dos moléculas, una de las cuales ingresaría al núcleo de la neurona, donde encuentra al ADN; a medida que más y más huntingtinas mutantes son escindidas, los segmentos moleculares resultantes se acumulan en el núcleo y comienzan a formar agregados al interior de éste (Figura 2). Sin embargo, el papel de estos agregados en la enfermedad de Huntington no está claro. Algunos especialistas creen que ellos matarían directamente a la célula, pero otros sostienen que son productos secundarios e inocuos de la huntingtina mutada (9).
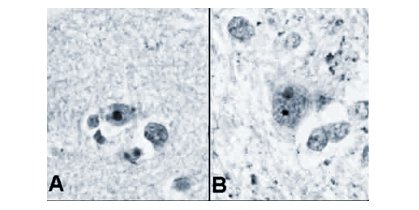 Tamaño completo
Tamaño completo Figura 2. Cortes histológicos de corteza cerebral (A) y núcleo caudado (B) de individuos que presentan la enfermedad de Huntington, en los que se observan cuerpos de inclusión intranuclear (11).
Incidencia y manejo
La prevalencia de la enfermedad de Huntington (EH) fluctúa entre 5 a 10 casos por 100.000 habitantes en países de Europa occidental, siendo algo menor en países del este asiático y en la población de raza negra. La incidencia anual varía entre 1 a 4 casos por millón de habitantes.
La edad de comienzo se sitúa entre los 30 y los 45 años, aunque puede producirse a partir de los dos años de edad. Los niños que desarrollan la enfermedad raramente alcanzan la edad adulta.
El diagnóstico se realiza mediante exploración física, TAC cerebral, resonancia magnética cerebral, PET cerebral y estudios del ADN (2). El diagnóstico genético se lleva a cabo a través del análisis de la historia familiar (árboles genealógicos).
El diagnóstico prenatal se realiza utilizando marcadores ligados al ADN, que no intentan pesquisar el gen para EH en el padre, sino que indican, en el marco de una familia con EH, si el feto ha adquirido el cromosoma 4 de un abuelo afectado o de uno que no lo está. La alternativa al diagnóstico prenatal consiste en realizar fecundación in vitro con screening preimplantacional del embrión e implantación de embriones no portadores.
No existe cura para la enfermedad de Huntington y no hay forma conocida de detener la progresión del trastorno; el tratamiento se orienta a reducir la progresión de la enfermedad y a maximizar la capacidad funcional del individuo, tanto como sea posible.
Los medicamentos varían de acuerdo con los síntomas; los bloqueadores de dopamina, como haloperidol o fenotiacina, pueden reducir los comportamientos y movimientos anormales; también se han utilizado reserpina y otros medicamentos, con éxitos variables, y drogas como la tetrabenazina y la amantidina, que se usan para tratar de controlar los movimientos adicionales. Hay evidencias que sugieren que la coenzima Q10 puede disminuir mínimamente el progreso de la enfermedad.
Las enfermedades siquiátricas, la depresión y el suicidio son comunes durante la enfermedad de Huntington, de modo que los médicos y las personas que cuidan al paciente deben monitorizar los síntomas y controlar el tratamiento adecuadamente. El tratamiento sintomático de la demencia es similar al de cualquier síndrome cerebral orgánico. Inicialmente, la señalización para recordar cosas y otras ayudas similares pueden mejorar la función de la memoria. Existe la necesidad de asistencia y supervisión progresivas, y se puede llegar a requerir cuidado durante las 24 horas del día (3).
El consejo genético en esta enfermedad es particularmente difícil, ya que es progresiva, incapacitante, tiene alto riesgo de transmisión hereditaria (50%) y, por el momento, no existe tratamiento preventivo ni curativo, sino únicamente paliativo. El consejero, sin querer, despierta diferentes reacciones, siendo las más frecuentes ansiedad, depresión y negación, por lo que debe establecer una buena relación con el paciente y su familia y programar varias sesiones conjuntas, que permitan a los familiares comprender y asimilar la información proporcionada.
La asesoría puede incluir el análisis de ADN a varios miembros de la familia. Como las posibilidades de que el hijo de una persona con la enfermedad de Huntington se vea afectado son muy altas, las personas con el trastorno pueden considerar la posibilidad de realizar adopción. También se puede considerar como una alternativa la reproducción asistida con selección de embriones no portadores, mediante análisis genético (2).
Desafortunadamente, debido al inicio tardío de la enfermedad, en la mayoría de los casos el paciente ya tiene hijos, e incluso nietos con la afección latente, cuando se hace el diagnóstico.
