Cursos
← vista completaPublicado el 1 de enero de 2005 | http://doi.org/10.5867/medwave.2005.01.3372
Fibrosis quística: aspectos genéticos
Cystic fibrosis: genetics
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
Descripción y características clínicas
La fibrosis quística (FQ) es un trastorno multisistémico hereditario, que se puede manifestar inicialmente de muchas formas diferentes (véase Tabla I). Sin embargo, las manifestaciones patológicas más invalidantes son de tipo respiratorio y digestivo.
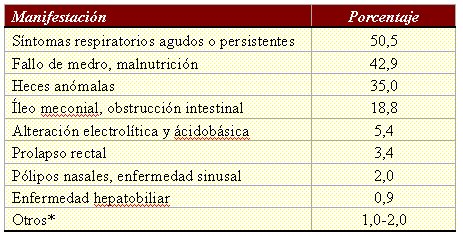 Tamaño completo
Tamaño completo Tabla I. Manifestaciones iniciales de más de 20.000 pacientes con fibrosis quística en Estados Unidos (* Incluye pseudotumor cerebral, azoospermia, exantema similar a la acrodermatitis, estados deficitarios de vitaminas, edema hipoproteinémico, hipoprotrombinemia con hemorragia, síndrome de tapón de meconio).
Los pulmones pueden parecer normales al momento del nacimiento, sin embargo, la acumulación posterior de secreciones mucosas conduce a infecciones recurrentes y a una destrucción progresiva del tejido alveolar. Además suele producirse un aumento en la resistencia de los vasos pulmonares, lo que puede conducir a hipertrofia del ventrículo derecho (cor pulmonale) .
La FQ también está asociada a excreción deficiente de enzimas pancreáticas, absorción intestinal reducida y a una consecuente mal nutrición. El 15% a 20% de los recién nacidos con FQ presentan obstrucción total del íleon por meconio (íleo meconial). El deterioro pancreático es progresivo y la muerte suele darse por complicaciones pulmonares (1, 3).
Además, la FQ está asociada con infertilidad masculina, debida a obstrucción o a ausencia congénita de los conductos deferentes y a una mayor excreción de sales en el sudor. Esto último por lo general no representa peligro de deshidratación para el paciente, excepto en ambientes calurosos. Sin embargo, sirve como prueba de diagnóstico para la enfermedad (1, 3).
Si bien la FQ es un trastorno que acorta la esperanza de vida, ésta puede llegar a superar los treinta años si se trata adecuadamente (4).
Bases genéticas
La FQ es una enfermedad autosómica recesiva producida por mutaciones en el gen CFTR ubicado en el brazo largo del cromosoma 7 (7q31) (5), el cual presenta gran expresión en las células epiteliales de las vías respiratorias, en el tubo digestivo (incluyendo el páncreas y el árbol biliar), en las glándulas sudoríparas y en el aparato urogenital. El producto de este gen corresponde a una proteína de 1480 aminoácidos llamada Regulador Transmembranoso de la FQ, la cual corresponde a un canal de iones selectivo para aniones, que además cumple funciones reguladoras que se alteran de forma variable por las distintas mutaciones (véase Figura 1).
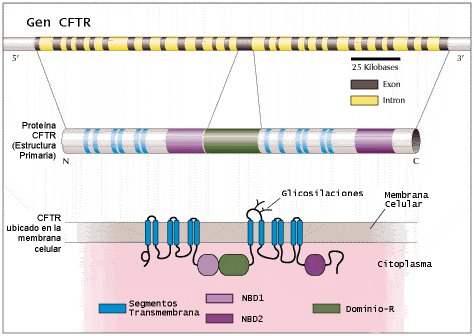 Tamaño completo
Tamaño completo Figura 1. Caracterización del gen CFTR humano y de su transcrito (2,16). El gen CFTR humano (arriba), identificado en 1989 en el brazo largo del cromosoma 7, consta de 27 exones que codifican para un polipéptido de 1480 aminoácidos (al centro). Esta proteína se ancla a la membrana celular por doce segmentos transmembrana (en azul). Estos segmentos forman un canal a través del cual pasa cloruro y, tal vez, agua. Dos dominios de unión a nucleótidos ( nucleotide binding domains , NBD1 y NBD2, en rosa y violeta, respectivamente) interaccionan con el ATP para suministrar la energía necesaria para la función de CFTR. El dominio R (en verde) tiene muchos sitios para su fosforilación por quinasas dependientes de cAMP. Este dominio está implicado en la regulación de las funciones de CFTR, tales como la conductancia de cloruro. La mutación más frecuente de CFTR, deltaF508, se localiza en la región NBD1. Esta región, así como NBD2 son especialmente susceptibles a las mutaciones.
La prevalencia de la FQ varía dependiendo del origen étnico de las poblaciones. Así, la FQ se detecta en aproximadamente 1 de cada 377 nacidos vivos en algunas zonas de Bretaña, 1 de cada 3.000 nacidos vivos en la población Caucásica de Norteamérica y del Norte de Europa, en 1 de cada 17.000 nacidos vivos norteamericanos de raza negra, y en 1 de cada 90.000 nacidos vivos en la población asiática de Hawaii. Sobre la base de la composición étnica de la población chilena (mezcla caucásico-amerindia), se ha estimado una incidencia de 1:4.000 nacidos vivos (6).
Si bien se han documentado más de 700 mutaciones que contribuyen al síndrome, sólo unas pocas se han descrito funcionalmente. Según los efectos que producen, éstas pueden ser agrupadas en cinco clases diferentes, que se describen a continuación (véase Figura 2).
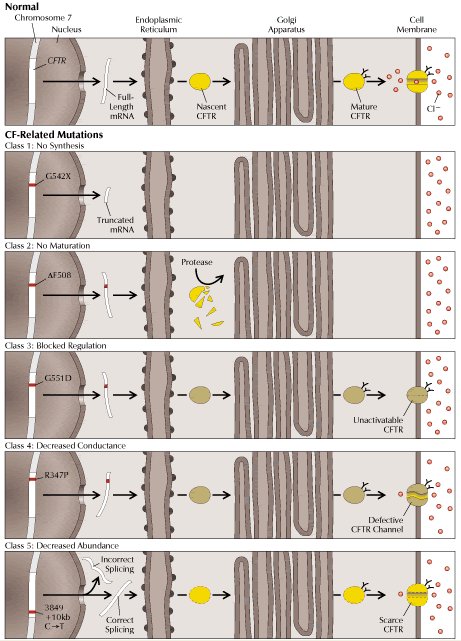 Tamaño completo
Tamaño completo Figura 2. Clases de mutaciones del gen CFTR (16). Cada una de las cinco clases de mutaciones altera la conductancia a cloruro de la membrana celular mediante un diferente mecanismo molecular. En condiciones normales (panel superior), el gen CFTR es transcrito en mRNA, el cual a su vez es traducido a proteína. Luego, la proteína sufre procesamiento post-traduccional previo a alcanzar su localización funcional definitiva. En los paneles inferiores vemos las distintas clases de mutaciones que implican patología (ver texto).
Las mutaciones clase I impiden la traducción normal, generando una proteína trunca, ya sea por anormalidades en los sitios de “splicing” , por corrimientos del marco de lectura ocasionados por deleciones o inserciones, o bien por mutaciones sin sentido. Esta clase está ejemplificada por G542X, en la cual un codón de término prematuro genera una proteína trunca no funcional.
Las mutaciones clase II alteran el procesamiento o el transporte adecuado de la proteína traducida hacia la membrana apical y resultan en una pérdida dramática de la expresión de CFTR en la superficie celular. Esta clase incluye a DF508, la que ocasiona un plegamiento incorrecto de CFTR que conduce a la proteólisis del péptido.
Las mutaciones clase III incluyen aquellas en que se ve alterada la regulación del canal, CFTR alcanza la superficie celular, pero su activación está impedida. Por ejemplo, G551D inactiva el dominio de unión a nucleótido NBD1.
Las mutaciones clase IV dan origen a canales con alteraciones en sus propiedades de conducción, por ejemplo conductancia a cloruro disminuida. El canal puede ser abierto, pero el paso de iones es más lento, esto último está ejemplificado por R347P, la que afecta un segmento de transmembrana.
Finalmente, en las mutaciones clase V sólo se ve alterada la abundancia de CFTR en la membrana celular. Esta clase está ejemplificada por una sustitución nucleotídica que aparentemente conduce a una mezcla de moléculas de mRNAs, algunas de las cuales sufre “splicing” incorrecto y las otras son procesadas normalmente.
Si bien esta clasificación es útil, no es completamente excluyente ya que algunas mutaciones pueden ser incluidas en más de una categoría. Las doce mutaciones más comunes encontradas en la población mundial se resumen en la Tabla II.
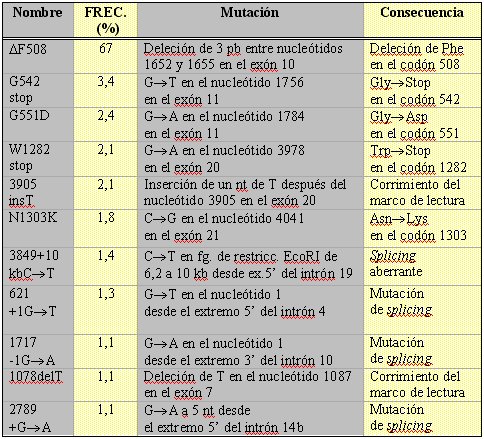 Tamaño completo
Tamaño completo Tabla II. Mutaciones más frecuentes del gen de la fibrosis quística (CFTR) (1,15) (pb = pares de bases, nt = nucleótido).
De éstas, la más frecuente, encontrada en alrededor de 70% de los pacientes caucásicos del Norte de Europa, corresponde a la deleción de un único aminoácido, una fenilalanina en la posición 508, en el primer dominio de unión a nucleótido (DF508). Esta mutación de clase II está asociada a una conductancia epitelial de cloruro activado por cAMP muy reducida, produciendo una forma severa de FQ. A 37 ºC la proteína mutante es procesada de manera incorrecta y no alcanza la membrana apical. Esta mutación es considerablemente menos frecuente en otras poblaciones, como las del Sur de Europa e Israel.
El resto de las mutaciones presentan una amplia dispersión. Así, ninguna de ellas tiene una prevalencia superior a unos pocos puntos porcentuales, salvo en poblaciones circunscritas. Por ejemplo, la mutación sin sentido W1282X se produce en 60% de los judíos asquenazíes con FQ. En el caso de la población chilena, hay estudios que sugieren que la mutación DF508 es efectivamente la más frecuente (7, 8), mientras que otros sugieren lo contrario (9). Estas diferencias se explican debido a que la tasa de detección alélica de estos estudios fue baja y el tamaño de las muestras fue relativamente pequeño, siendo difícil llegar a conclusiones definitivas.
La relación entre genotipo y fenotipo en la FQ es sumamente compleja. Las mutaciones consideradas “graves” como DF508, se asocian casi siempre a insuficiencia cardíaca. Existen otras mutaciones, como 3849 + 10kb C®T, que se asocian a concentraciones normales de cloruro en el sudor (10). El genotipo permite predecir el fenotipo pancreático, sin embargo no permite predecir la gravedad de la enfermedad pulmonar ni la presencia de hepatopatía. Este hecho sugiere un componente ambiental o adquirido de la disfunción orgánica, o bien, la presencia de genes modificadores pertenecientes al genoma residual, que aún no se han identificado y contribuyen al fenotipo de la FQ dándole expresividad variable.
Patogénesis
Existen cuatro caracteres patológicos que tienen una importancia fundamental:
- La incapacidad de aclarar las secreciones mucosas.
- La escasez de agua en las secreciones de moco.
- El elevado contenido de sal en el sudor.
- La infección crónica limitada al aparato respiratorio.
La relación entre estas características no se aclaró hasta la década de los 80, en que se descubrió que los pacientes tienen una alteración en la conductancia de cloruro en las membranas epiteliales, efecto que posteriormente se atribuyó a la disfunción del producto del gen CFTR (11). El mecanismo fisiopatológico propuesto para el epitelio de las vías respiratorias, implica incapacidad de secretar sal y, secundariamente, incapacidad de secretar agua por una reabsorción excesiva de ambas sustancias (véase figura 3).
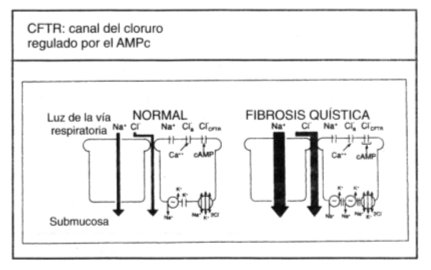 Tamaño completo
Tamaño completo Figura 3. Disfunción del epitelio respiratorio en FQ (2). Flujo neto de iones a través de los epitelios de las vías respiratorias en sujetos normales y en los que padecen fibrosis quística (FQ) en condiciones basales (flechas gruesas). Debido a que el agua sigue a los desplazamientos de la sal, el flujo neto de agua previsto partirá desde la luz de las vías respiratorias hasta la submucosa y será mayor a través de los epitelios con FQ, provocando resecación de las secreciones.
Se cree que como resultado final de estas alteraciones se produce escasez de agua para hidratar las secreciones de la superficie de las vías respiratorias. Las secreciones desecadas se tornan más viscosas y elásticas (gomosas), y son más difíciles de eliminar por el sistema mucociliar y por otros mecanismos. Estas secreciones se retienen y obstruyen las vías respiratorias, comenzando por aquellas de menor calibre, como los bronquiolos. Así, la primera alteración fisiológica perceptible de la respiración es la obstrucción del flujo aéreo en las vías respiratorias pequeñas (12).
Es factible que ocurra un proceso fisiopatológico similar en el árbol pancreático y biliar y también en los conductos deferentes, que origine desecación de las secreciones serosas y obstrucción. En los conductos excretores de las glándulas sudoríparas normalmente ocurre reabsorción de cloruro y, por ende, de Na+; en los pacientes con fibrosis quística, al estar alterado el gen CFTR, no se pueden efectuar dichas reabsorciones y, como consecuencia, se elevan los niveles de cloruro y de sodio en el sudor.
Tal como se puede ver en la figura 4, normalmente (arriba), la glándula recibe el agua y electrolitos que excreta desde los vasos sanguíneos por difusión, reabsorbiendo el cloruro a través del canal llamado “Regulador Transmembranoso de la FQ” (CFTR). En la FQ (abajo) el canal está alterado o ausente. Los esfuerzos para entender su electrofisiología se complican por el hecho de que normalmente CFTR provoca un aumento de la conductancia al cloruro del canal ORCC (outwardly-rectifying chloride channel) y, a la vez, un decremento de la conductancia al sodio del canal de sodio epitelial (ENAC). Por lo tanto, la disfunción de CFTR va acompañada por un incremento del paso de sodio y de un decremento del paso de cloruro. La influencia de CFTR sobre un canal de cloruro dependiente de calcio todavía no se conoce. Para simplificar el esquema, todas las conductancias se muestran en un único tipo celular, pero los datos disponibles provienen de varios tejidos diferentes; de hecho, en el pulmón y el páncreas CFTR normalmente posibilita la secreción de cloruro y no su reabsorción (véase Figura 4).
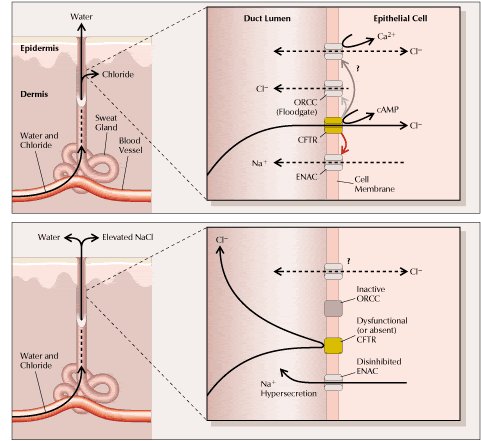 Tamaño completo
Tamaño completo Figura 4. Excreción anormal de sal en el sudor en los pacientes con FQ (1,16). El defecto biomolecular primario subyacente a la FQ es una anomalía en el transporte de cloruro en la membrana apical de las células epiteliales secretoras, como se esquematiza aquí para una glándula sudorípara (ver más detalles en el texto).
La infección crónica en la FQ se limita a los espacios endobronquiales de las vías respiratorias. La explicación más probable de la infección es una secuencia de sucesos que comienzan con la dificultad para expulsar con rapidez las bacterias inhaladas, seguida de la colonización persistente y de una respuesta inflamatoria de las paredes de las vías respiratorias. Con la enfermedad pulmonar avanzada, la enfermedad puede extenderse al parénquima pulmonar peribronquial.
Diagnóstico y consejo genético
Procedimientos de hibridación con sondas específicas para las 30 mutaciones más frecuentes han permitido determinar el genotipo del 80% al 90% de los estadounidenses con FQ, pero como las frecuencias alélicas de las mutaciones varían según la población en estudio, en general no es posible extrapolar estos resultados a otras poblaciones. Si se aumenta el número de sondas hasta 70, la identificación de la mutación mejora sólo en un porcentaje reducido. Debido a esta gran heterogeneidad genética, no es posible identificar a todos los niños con FQ mediante las pruebas de DNA que se practican en la actualidad, por lo cual el diagnóstico en la mayoría de los casos se basa fundamentalmente en criterios clínicos y en la medición de cloruro en el sudor (2, 3).
El consejo genético se entrega a los padres de niños con FQ, explicándoles la probabilidad de que sus futuros hijos estén afectados o sean portadores, permitiendo que la pareja planifique su natalidad informadamente. En cuanto a la detección de portadores, ésta se realiza mediante exámenes genéticos, y se centra principalmente en aquellos familiares de enfermos o portadores conocidos y en sus respectivas parejas, puesto que es poco práctico aplicar este tipo de exámenes genéticos a la totalidad de la población. En caso de que el individuo resulte portador, se procede a informarlo acerca de su condición. Se han implementado numerosos programas con este objetivo, por ejemplo, el existente en Inglaterra desde el año 1993 en el Royal Manchester Children’s Hospital (13).
El hecho de que la FQ sea una enfermedad autosómica recesiva, hace que una pareja de portadores tenga un cuarto de posibilidades de tener un hijo afectado, un medio de tener un hijo sano portador, y un cuarto de tener un hijo sano no portador. Sin embargo, por la gran heterogeneidad genética y expresividad variable de esta enfermedad, muchas veces el consejo genético se hace complejo (14), ya que no es posible predecir con exactitud el grado de manifestación fenotípica de la enfermedad, el que puede ser muy variable.
A pesar de que la mayoría de los hombres enfermos de FQ son infértiles, no sucede necesariamente lo mismo con las mujeres, las que pueden concebir hijos al llegar a su edad reproductiva. En estos casos, la decisión de tener hijos recae sobre cada paciente, por lo que es bueno informarlas sobre los riesgos e implicancias de su condición, tanto para sí mismas como para la nueva vida que está por nacer.
