Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso Internacional de Diabetes del Niño y del Adolescente, organizado por la Asociación Latinoamericana de Diabetes entre los días 19 al 21 de abril de 2006. Directora: Dra. Gloria López.
Introducción
La enfermedad cardiovascular aterosclerótica constituye la mayor causa de morbimortalidad de los pacientes portadores de diabetes mellitus; a su vez, la diabetes y la dislipidemia constituyen factores de riesgo primordiales en el desarrollo de la enfermedad cardiovascular aterosclerótica.
Dislipidemia en diabetes mellitus tipo 1
La dislipidemia de pacientes con diabetes mellitus tipo1 (DM1) es expresión de alteraciones en el metabolismo de las lipoproteínas ricas en triglicéridos. Frente a una disminución o ausencia de insulina, como ocurre en la DM1, la lipasa lipoproteica y la lipasa hepática, las dos enzimas claves en este metabolismo, están disminuidas, conduciendo a una reducción del catabolismo o degradación de las lipoproteínas ricas en triglicéridos, principalmente VLDL (Very Low Density Lipoprotein), IDL (Intermediate Density Lipoprotein) y quilomicrones, en el momento postprandial. El perfil que se observa en estos pacientes es aumento de VLDL, IDL, con colesterol HDL (High Density Lipoprotein) en general normal.
Dislipidemia en diabetes mellitus tipo 2
En la diabetes tipo 2 (DM2) la insulina está aumentada como respuesta a la resistencia a ésta, lo que origina un flujo importante de ácidos grasos desde el adipocito hacia el hígado, donde son empaquetados en la forma de triglicéridos y excretados a la circulación como VLDL. Además, la resistencia a la insulina reduciría la actividad de la lipasa lipoproteica y de la lipasa hepática, disminuyendo así el catabolismo de las lipoproteínas ricas en triglicéridos. En suma, hay sobresíntesis de triglicéridos a nivel hepático, junto a una disminución de su catabolismo. La dislipidemia de estos pacientes se caracteriza por aumento de VLDL, mayor que el que se observa en la DM1, aumento de IDL y disminución del colesterol HDL asociado al aumento de los triglicéridos. Por este motivo las terapias hipolipemiantes, dirigidas a bajar los niveles de triglicéridos, producen generalmente elevación de HDL.
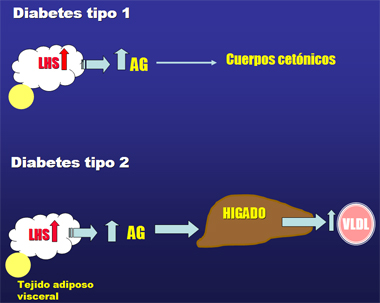
A diferencia de lo que se observa en la DM2, en que debido a la resistencia a insulina, hay mayor secreción de ácidos grasos por parte del adipocito los cuales llegan al hígado, donde son empaquetados y enviados a la circulación como partículas de VLDL (Fig. 1), en la DM1 los ácidos grasos en exceso van principalmente hacia la síntesis de cuerpos cetónicos (Fig. 1).
;;;;Figura 1. Alteraciones en el metabolismo de los lípidos en DM1 y DM2
En la DM2 no sólo aumenta la cantidad de partículas de VLDL, también varía la calidad de esta lipoproteína, caracterizada por la presencia de moléculas grandes y sobrecargadas en triglicéridos. Además, hay una prevalencia de VLDL pequeñas y ricas en colesterol denominadas beta VLDL. La importancia de este cambio cualitativo en cuanto a tamaño de la partícula es crucial: frente a un aumento de la permeabilidad del endotelio no son las grandes VLDL las que penetran la pared vascular, sino las VLDL pequeñas. Por lo tanto, estas partículas de menor tamaño se pueden asociar con la gravedad y progresión de la ateroesclerosis.
Otro aspecto cualitativo a considerar es la composición proteica de las VLDL. Si la VLDL contiene Apo C III, un inhibidor importante de la enzima lipoproteinlipasa, la lipólisis estará diminuida aumentando así las partículas pequeñas más susceptibles de penetrar la pared vascular, característica que las vuelve más aterogénicas. Así, el tamaño y la composición de las lipoproteínas son los principales factores determinantes de su aterogenicidad.
IDL
El metabolismo de las IDL se altera tanto en la diabetes mellitus tipo 1 como en la tipo 2. En la DM2 hay un aumento importante de IDL, provocado por un incremento en sus precursoras, las VLDL; hay hipertrigliceridemia, con predominio de partículas grandes de VLDL, las que, por lipólisis, originan grandes cantidades de IDL, que son reconocidas por receptores hepáticos específicos para ellas (Fig. 2). En la DM1 habría una falla en el catabolismo de estas partículas: la lipasa hepática (LH), activada por insulina, disminuye frente a la ausencia de esta hormona, disminuyendo así el catabolismo de las IDL (Fig. 2).
La IDL está aumentada, tanto en la DM1 como en la DM2. Al medir el colesterol de IDL (col-IDL), como expresión de cantidad de esta lipoproteína se observan niveles significativamente superiores, tanto en la DM1 como en DM2, comparados con los valores de sujetos controles no diabéticos (Tabla I).
;;;;Tabla I. Valores medios y percentiles de colesterol IDL en diabéticos
Las LDL constituye el objetivo primario en el tratamiento de la dislipidemia en pacientes diabéticos. El ATP III (Adult Treatment Panel III) ha señalado a la LDL como el blanco esencial de las terapias farmacológicas y no farmacológicas; incluso, se ha establecido en pacientes diabéticos, que la meta es lograr valores de colesterol LDL inferiores a 70 mg/dl.
En los pacientes con diabetes las LDL están dentro de rangos normales, e incluso disminuidos, pero son generalmente pequeñas y densas, altamente aterogénicas. Las LDL tienen, en sus dominios ligandos para receptores, sitios importantes de unión a glucosa, transformándose en LDL glicadas, las cuales presentan menor afinidad por su receptor y se oxidan con mayor facilidad. Las LDL oxidadas constituyen ligandos esenciales para los receptores scanvenger de los macrófagos, que al captar e internalizarlas, en un proceso no regulado, acumulan colesterol, transformandose en las llamadas células espumosas. En suma, hay tres modificaciones importantes que aumentan la aterogénesis de las LDL en el diabético: LDL pequeña y densa, LDL oxidada y LDL glicadas.
La LDL pequeña y densa constituyen un factor de riesgo de infarto: el riesgo aumenta hasta tres veces en presencia de esta lipoproteína y entre un 40% y 50% de los hombres con enfermedad coronaria presentan niveles altos de LDL pequeñas y densas. Actualmente es posible medir las LDL pequeñas y densas de manera indirecta, mediante el cuociente colesterol/ApoB.
Hay varias causas que explican la generación de LDL pequeñas y densas en diabéticos (Fig. 3). La primera es el tipo de lipoproteína precursora. Si la VLDL del cual se origina es muy rica en triglicéridos lo más probable es que lleve a la formación de una partícula de LDL densa y pequeña, por la transferencia de colesterol y triglicéridos que realiza la CETP (Cholesterol Ester Transfer Protein), situación que se da preferentemente en DM-1. La otra causa es cuando las VLDL son muy ricas en triglicéridos, los transfieren a las LDL, las que devuelven a las VLDL ese contenido de hidrofobicidad, en forma de colesterol. La LDL se enriquece en triglicéridos, pierde colesterol y de esa manera se hace más pequeña y densa; situación que se presenta en ambos tipos de diabetes.
Se había citado como ejemplo que las VLDL ricas en Apo CIII contienen mayor cantidad de triglicéridos, debido a que esta apolipoproteina es un inhibidor de la enzima liporprotienlipasa. Estas VLDL dan origen a una LDL que no es bien reconocida por los receptores del hígado, permaneciendo mas tiempo en circulación. Esto hace que penetren la capa endotelial, quedando vulnerables al ataque oxidativo y transformándose en LDL oxidadas, las cuales son captadas rápidamente por los receptores scavenger de los macrófagos que acumulan el colesterol que proviene de las LDL densas y pequeñas, transformándose en células espumosas.
HDL
El estudio Framingham demostró que con sólo aumentar 1 mg/dl el colesterol de HDL (col-HDL), disminuye el riesgo cardiovascular en 2% en los hombres y en 3% en las mujeres. Esta lipoproteína ejerce un rol antiaterogénico y se la considera por ello protectora de cardiopatía coronaria. La HDL participa en la retirada del colesterol desde los macrófagos: el colesterol libre fluye por un proceso mediado por el transportador ABCA1; las partículas de HDL nacientes (pre HDL) captan preferencialmente este colesterol y una vez esterificado lo ingresan al hígado a través de un proceso de captación selectiva de los esteres de colesterol mediado por un receptor scavengerlocalizado en la membrana de los hepatocitos.
Transporte reverso de colesterol (Fuente: Chinetti G. Nat Med 2001; 7(1): 53-58)
En los diabéticos, tipo 2 como tipo 1, aumenta la proteína de transferencia de los ésteres de colesterol CETP (Fig. 5); ésta promueve la transferencia de triglicéridos desde las VLDL hacia las HDL a cambio de esto las VLDL reciben colesterol esterificado. La HDL se transforma así en una partícula rica en triglicéridos y pobre en colesterol, que se cataboliza con más rapidez, disminuyendo sus niveles en circulación, lo que conlleva a un ineficiente transporte reverso de colesterol.
Es importante considerar que la HDL es una partícula heterogénea en su composición proteica. Habitualmente se mide el colesterol de HDL, es decir, la cantidad de colesterol que contiene la partícula. Sin embargo se han descrito dos tipos de lipopartícula de HDL: una que contiene sólo ApoA-I (LpA-I) y otra que contiene Apo A-I y Apo A-II (LpA-I:A-II). El ligando para el transportador que media la retirada de colesterol libre desde el macrófago es el Apo A-I, del mismo modo el apo A-I es el activador esencial para la transformación del colesterol libre en colesterol esterificado y convertir así las partículas nacientes en HDL maduras; además, Apo A-I es el ligando para que los receptores del hígado capten selectivamente el colesterol esterificado. Apo A-II desplazaría al Apo A-I de la superficie de la partícula de HDL y restaría eficacia al proceso. Es posible plantear que un aumento de HDL, a expensas de partículas LpAI aumentaría la eficiencia del transporte reverso de colesterol; por lo tanto, la cantidad de colesterol HDL tendría un valor relativo, frente a la capacidad realmente antiaterogénica de la partícula.
En un estudio colaborativo, para lo cual se produjeron en nuestro laboratorio anticuerpos monoclonales específicos para medir estas partículas con test de ELISA, se observó que los sujetos diabéticos tenían valores menores de Lp A-I, que los sujetos controles (Fig. 6).
Concentraciones de Lp A-I y Lp A-I;A-II en diabéticos tipo 2 y sujetos controles (* versus controles, p menor de 0,005).
En otro estudio dirigido por Asenjo y colaboradores, en niños y jóvenes con diabetes tipo 1, en que no se encontró una disminución o diferencia significativa en los valores de HDL, los que incluso fueron más altos en los niños diabéticos tipo 1, que en los controles sanos. Se observó un predominio de las partículas de Lp A-I encontrándose diferencias en la relación Lp A-I/Lp A-II, en comparación con los controles no diabéticos.
También se evaluó el tamaño de la partícula de HDL en los sujetos diabéticos y en los controles, mediante la relación colesterol HDL/ApoA-I, que permite estimar el tamaño aproximado de la partícula. En los sujetos diabéticos se observó mayor cantidad de partículas grandes que en los controles. Cuando se estimó el tamaño de las partículas LDL, por el mismo método de colesterol sobre Apo B, componente proteico de la LDL, se encontró mayor cantidad de partículas pequeñas y densas en los diabéticos, mientras en los controles predominaron las partículas grandes y livianas. En este estudio se demostró que, aunque los pacientes diabéticos tengan valores normales de HDL, puede haber diferencias cualitativas importantes.
Además se midió en suero la glicación de las HDL con anticuerpos monoclonales dirigidos contra formas glicadas de esta lipoproteína, usando test de ELISA. En los sujetos diabéticos se observó mayor glicación de HDL, en comparación a los sujetos controles no diabéticos. Esta reactividad específica, medida como glicación, también es más frecuente en los pacientes diabéticos.
En un estudio en colaboración con el Profesor Fruchart se separó mediante cromatografía de inmunoafinidad las formas glicadas y no glicadas de partículas LpAI. Se cargaron macrófagos con colesterol tritiado y se determinó la capacidad que tenían las formas glicadas y no glicadas para ejercer eflujo de colesterol desde estos macrófagos. La forma glicada ejerció prácticamente el 50% de la capacidad para hacer efluir colesterol desde macrófagos, comparado con la forma no glicada.
En otro estudio, se midió la oxidabilidad de HDL aislada de pacientes diabéticos y de controles. Las TBARS (especies reactivas al ácido tiobarbitúrico) mostraron un grado mayor de oxidación de HDL en diabéticos con respecto a los no diabéticos, lo cual se corroboró con el lag phase, que mide el grado de protección del HDL frente a la oxidación. Se observó que en los pacientes diabéticos la HDL está menos protegida frente a la oxidación, posiblemente atribuible a una baja actividad de la enzima paraoxonasa asociada a la HDL.
Otro aspecto interesante de analizar es la dislipidemia típica del diabético tipo 2 y del síndrome metabólico: hipertrigliceridemia asociada a HDL bajo y predominio de partículas densas y pequeñas de LDL. Está claro que la hipertrigliceridemia condiciona una baja HDL y que favorece el desplazamiento de las partículas LDL grandes y livianas hacia partículas densas y pequeñas, más aterogénicas. Aumentan las partículas ricas en triglicéridos, por baja lipólisis, secundario a la presencia del inhibidor de la lipasa, el Apo C III. Al mismo tiempo se observa una disminución de la HDL, principalmente a expensas de la partícula Lp A-I, que es la más eficaz para retirar colesterol desde el macrófago.
Agonistas de PPAR alfa como fibratos o de PPAR gamma como las glitazonas inducen la expresión del gen de la lipoproteinlipasa. Los PPARs son factores de transcripción que una vez activados se unen a la región promotora de los genes blancos en este caso, de la lipoproteinlipasa aumentando su expresión. Frente a una mayor cantidad de enzima se degradan más eficazmente los triglicéridos de las VLDL y aumentan los niveles de HDL como consecuencia de la liberación de exceso de superficie remanentes resultante de la hidrólisis de las VLDL (Fig. 7). Bajo terapia con fibratos o glitazonas, los triglicéridos disminuyen generalmente asociado a un aumento en los niveles de colesterol HDL.
;;;;Figura 7. PPAR alfa y gamma aumentan la expresión del gen de lipoproteínlipasa (LPL)
Activadores de PPAR alfa, tipo fibratos, también desplazan las VLDL grandes, ricas en Apo CIII; que por transferencia mediada por CETP van a enriquecer a las LDL con triglicéridos, con lo que se tornan más densas y pequeñas, más susceptibles de ser captadas por los macrófagos y, por ende, más aterogénicas. El activador de PPAR alfa desplazaría esta partícula rica en Apo CIII y en triglicéridos, hacia una partícula pobre en Apo CIII y en triglicéridos. Como consecuencia un menor desplazamiento de triglicéridos ocurre hacia la LDL permaneciendo así grande y liviana, fácilmente captadas por los receptores del hígado y de los tejidos periféricos.
Finalmente, con activadores de PPAR alfa aumenta la expresión de los genes de Apo A-I y Apo A-II, incrementando las partículas de HDL; se induce el gen de la lipoproteinlipasa y se reprime el gen del Apo CIII, aumentando la lipólisis de las lipoproteinas ricas en triglicéridos.
Aterosclerosis
El evento inicial de la ateroesclerosis lo constituye la disfunción endotelial; como consecuencia de ello aumenta la permeabilidad a monocitos circulantes y a LDL nativas, los que ingresan a la pared vascular. Una vez en el interior, el monocito se transforma en macrófago y la LDL nativa es oxidada. El macrófago expresa receptores scavenger que capta con alta afinidad las LDL oxidadas y acumula colesterol en forma no regulada, conduciendo a la formación de células espumosas, que van a constituir la estría lipídica. La estría, impregnada en células espumosas secreta gran cantidad de proteínas proinflamatorias que van a desencadenar un estado inflamatorio crónico. El rompimiento de una placa inestable desencadenaría el evento coronario.
Finalmente en la diabetes, la hiperglicemia crónica va a llevar a una acumulación de proteínas glicadas que van a ser reconocidas por los receptores RAGE (Receptor for Advanced Glycation EndProducts), o receptores de productos de glicosilación avanzada. Al activarse estos receptores se produce liberación de citoquinas (IL-1) y factor de necrosis tumoral alfa (TNF alfa), que inducen a la célula endotelial a producir moléculas de adhesión promoviendo que el monocito se adhiera al endotelio; proteínas quimiotácticas a monocitos que favorecen el ingreso del monocito a la pared vascular y proteínas de diferenciación, que van a transformar este monocito en macrófago. La LDL, que ingresa por el aumento en la permeabilidad vascular, se va a oxidar y va a ser captada por los macrófagos. El macrófago activado libera TNF alfa e interleuquinas, lo que desencadenará un estado proinflamatorio. Finalmente se activará el proceso de coagulación, la producción de factores de crecimiento y la proliferación de células musculares lisas (Fig. 8).
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
 Tamaño completo
Tamaño completo 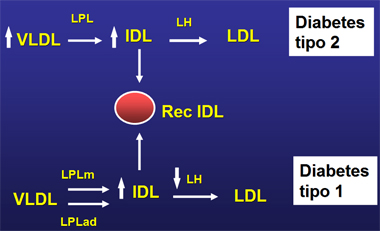 Tamaño completo
Tamaño completo 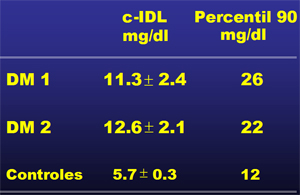 Tamaño completo
Tamaño completo 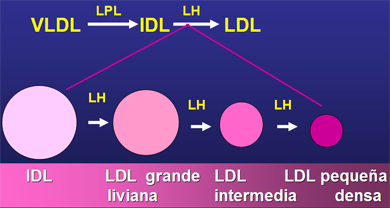 Tamaño completo
Tamaño completo 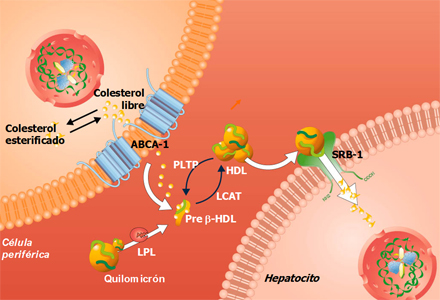 Tamaño completo
Tamaño completo 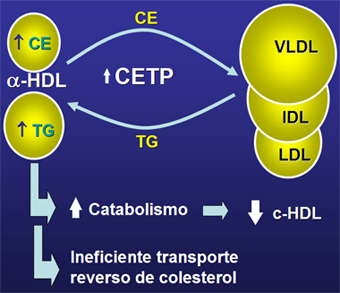 Tamaño completo
Tamaño completo 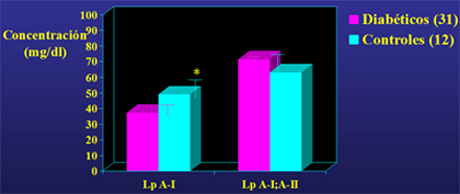 Tamaño completo
Tamaño completo 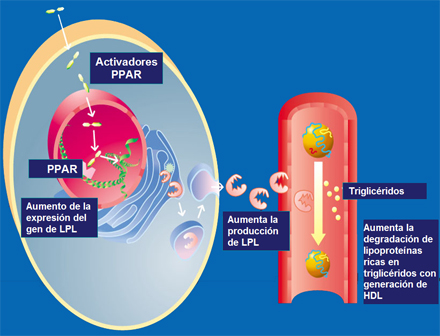 Tamaño completo
Tamaño completo 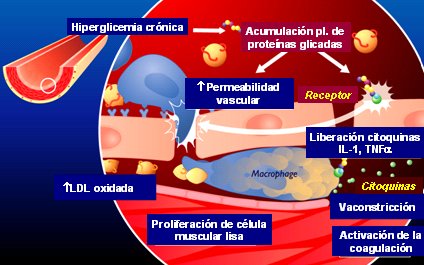 Tamaño completo
Tamaño completo 