Cursos
← vista completaPublicado el 1 de mayo de 2007 | http://doi.org/10.5867/medwave.2007.04.3547
Rol de los estudios citogenéticos y de genética molecular en el diagnóstico de tumores pediátricos de células pequeñas redondas y azules
Role of cytogenetics and molecular genetics in the diagnosis of small blue round cell pediatric tumors
Resumen
Este texto corresponde a un trabajo de revisión preparado por sus autores en el desarrollo del Curso y Seminarios de Oncología Básica, realizado por el Centro de Oncología Preventiva y la Escuela de Postgrado de la Facultad de Medicina de la Universidad de Chile entre abril y agosto de 2006. El Director del Curso es el Dr. José Manuel Ojeda.
Introducción
El término “tumores de células pequeñas redondas y azules” designa a un grupo heterogéneo de neoplasias malignas, que típicamente se presentan en niños y adultos jóvenes, cuyo aspecto histológico es similar: se caracterizan por una proliferación monótona de células indiferenciadas, de tamaño relativamente pequeño, con escaso citoplasma y núcleo hipercromático, pudiendo ser indistinguibles a la microscopía óptica, por lo que su diagnóstico preciso es a menudo un desafío para el patólogo. Si bien su apariencia morfológica es uniforme, es imprescindible su correcta distinción, debido a que el pronóstico y tratamiento difieren según el tipo de tumor y algunas terapias incluyen regímenes intensivos de quimio y/o radioterapia, con sus consecuentes riesgos. Este grupo de tumores incluye:
- tumores de la familia de Ewing
- neuroblastoma
- rabdomiosarcoma
- linfoma
- tumor desmoplástico de células pequeñas redondas
- osteosarcoma de células pequeñas y
- condrosarcoma mesenquimal.
El primer gran avance para lograr un diagnóstico más preciso fue el desarrollo de los anticuerpos monoclonales, que permitió, a través de técnicas de inmunohistoquímica, separar a la mayoría de los linfomas y al neuroblastoma de este grupo. El diagnóstico de los tumores de la familia de Ewing se hacía inicialmente por exclusión de los otros miembros, pero esto comenzó a cambiar cuando los estudios citogenéticos permitieron describir translocaciones específicas y patognomónicas para los tumores de la familia de Ewing y luego para otros tumores de células pequeñas redondas, como el rabdomiosarcoma alveolar y el tumor desmoplástico de células pequeñas redondas. Actualmente, la posibilidad de clasificar estos tumores por su perfil genético se ha acelerado con el uso de la tecnología de los microarrays, que se vislumbra como una poderosa herramienta a futuro.
Epidemiología
Los datos epidemiológicos nacionales muestran la importancia del problema y se correlacionan cercanamente con los datos internacionales. En Chile, la leucemia, principalmente la leucemia linfoblástica aguda (LLA) es la neoplasia maligna más frecuente en menores de 15 años, seguida por los tumores del sistema nervioso central, los linfomas y en cuarto lugar, dentro de los tumores sólidos, los sarcomas de partes blandas (SPB), cuyo principal representante es el rabdomiosarcoma (Tabla I).
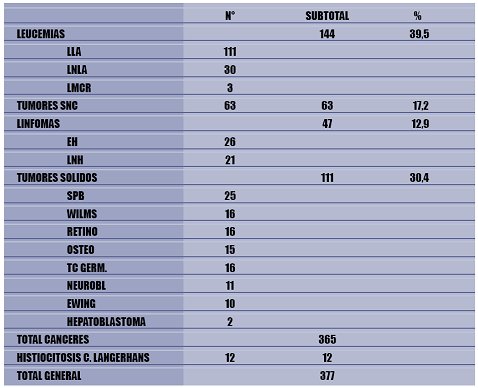 Tamaño completo
Tamaño completo Tabla I. Estimación de casos nuevos por año a base del promedio de casos estudiados en 10 años, en el período 1987-1997, en la Red del PINDA
Métodos citogenéticos y de genética molecular
Los cambios genéticos de valor en el diagnóstico diferencial de tumores de células pequeñas redondas incluyen las translocaciones cromosómicas, con la consiguiente fusión de genes (tumores de la familia de Ewing, rabdomiosarcoma alveolar, tumor desmoplástico de células pequeñas redondas y linfomas), número de copias de DNA y amplificación de genes (neuroblastoma). Las técnicas más comúnmente usadas para detectar fusión de genes son FISH y RT-PCR. Los estudios moleculares, por su disponibilidad y dificultades técnicas, se pueden realizar sólo en algunos casos. En general se requiere una muestra representativa mínima de 100 mg de tejido tumoral viable congelado (1, 2).
Análisis citogenético convencional: muestra translocaciones y otras anomalías estructurales, como aberraciones numéricas. Es necesario tejido tumoral viable. Pueden existir cariotipos falsamente normales, relacionados con condiciones de cultivo celular subóptimas, muestra de tejido necrótico o no representativa, enmascaramiento por sobrecrecimiento de fibroblastos estromales y presencia de alteraciones submicroscópicas.
Hibridización in situ fluorescente o cromogénica (FISH o CISH): a través de tinción de cromosomas o con sondas para puntos específicos de quiebre es posible detectar translocaciones y con sondas gen específicas, identificar amplificación de genes. Ambas técnicas se pueden realizar en tejido incluido en parafina. El FISH multicolor o cariotipo espectral es capaz de revelar con más precisión alteraciones cromosómicas complejas (Fig. 1).
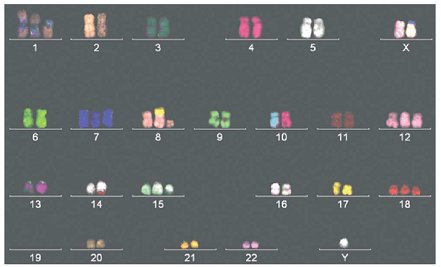 Tamaño completo
Tamaño completo Figura 1. FISH multicolor o cariotipo espectral
Hibridización genómica comparativa (CGH): muestra cambios del número de copias de secuencias de DNA (pérdidas, ganancias y amplificaciones), pero no detecta translocaciones balanceadas. Puede utilizarse tanto tejido fresco como fijado en formalina y su sensibilidad es afectada por el porcentaje de células tumorales de la muestra.
Métodos basados en PCR: identifica rearreglos genéticos y translocaciones con fusión de genes. Sus ventajas son la rapidez, efectividad y alta sensibilidad, lo que permite utilizar pequeñas cantidades de tejido, siendo útil en el monitoreo de enfermedad residual mínima y micrometástasis. Es importante minimizar al máximo la posibilidad de contaminación y asegurar la calidad de la muestra, debiendo ser rápidamente inhibida la degradación del DNA mediante congelación.
Métodos basados en microarray: han permitido la clasificación de los tumores a base de su perfil de expresión genética, por su capacidad para analizar simultáneamente la expresión de miles de genes y a través del análisis de cluster, identificar grupos pronósticos.
Tumores de la familia de Ewing
Los tumores de la familia de Ewing incluyen al sarcoma de Ewing, el tumor neuroectodérmico primitivo o PNET y el tumor de Askin. En su diagnóstico son de utilidad las técnicas de inmunohistoquímica, que muestran un alto nivel de expresión de CD99 (3), anticuerpo monoclonal que detecta un epítope de la glicoproteína de superficie MIC2; sin embargo, esta reacción no es específica y se ha visto positividad en otros tipos de tumores de células pequeñas redondas.
Las alteraciones citogenéticas en cambio, son específicas, caracterizándose por translocaciones que comprometen al gen EWS y a uno de varios miembros de la familia de factores de transcripción ETS, las que se pueden identificar mediante técnicas de RT-PCR o FISH (4 y 5). En 85 a 90% de estos tumores se detecta la translocación t(11;22) (q24;q12), como se muestra en las figuras 2 y 3, lo que genera un transcripto de fusión quimérico de EWSR1 y FLI1 (6). Una minoría de los tumores de Ewing presentan variantes de translocaciones en que EWS se fusiona con otros miembros de la familia de genes ETS: ERG, ETV1, E1AF, FEV y ZSG (7, 8, 9 y 10). La fusión clásica de transcriptos se asocia con mejor pronóstico, en comparación con las otras variantes (11).
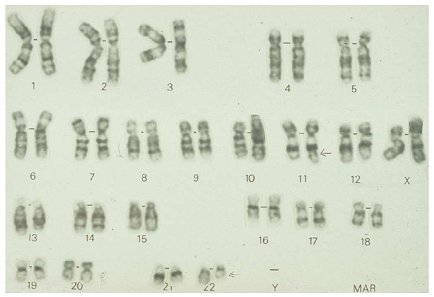 Tamaño completo
Tamaño completo Figura 2. Cariotipo de células tumorales de un paciente con sarcoma de Ewing. Las flechas muestran la translocación clásica t(11;22)
 Tamaño completo
Tamaño completo Figura 3. FISH con 2 sondas (verde y violeta) alrededor del gen EWS (flecha roja). La separación de las señales verde y violeta (flechas negras) indica la translocación t(11;22)
Neuroblastoma
Es el tumor sólido más común en el lactante. Desde el punto de vista inmunohistoquímico, expresa marcadores neurales como sinaptofisina, neurofilamentos y enolasa neuroespecífica, siendo todos inespecíficos. La microscopía electrónica muestra procesos neuríticos con gránulos neurosecretores y microtúbulos, hallazgos ultraestructurales que son patognomónicos de neuroblastoma (12).
No se han descrito anomalías citogenéticas diagnósticas consistentes, sin embargo, se han identificado alteraciones genéticas de significación pronóstica: la deleción del brazo corto del cromosoma 1, que es la más característica (13); la amplificación del oncogen MYCN (Fig. 4), que se manifiesta como dobles diminutos o zonas de tinción homogénea, que se ha correlacionado con rápida progresión y peor pronóstico (14); la hiperdiploidía, que ha mostrado mejor respuesta al tratamiento que los tumores diploides (15); la ganancia del cromosoma 17q, que también se ha asociado con un pronóstico adverso (16) y la expresión de altos niveles de la familia de receptores de neurotropinas TrkA, que ser asocia a una evolución favorable.
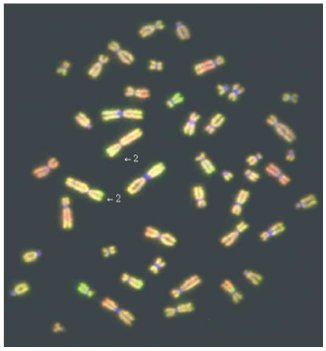 Tamaño completo
Tamaño completo Figura 4. Neuroblastoma de alto grado estudiado con CGH. Verde señala ganancia y amplificación de alto nivel; rojo, pérdida de número de copias de secuencias de DNA. Un claro incremento del número de copias en el brazo corto del cromosoma 2, indica amplificación de MYCN
La expresión de genes involucrados en la biosíntesis de catecolaminas, con coexpresión de tirosina hidroxilasa y dopa decarboxilasa ha mostrado ser una característica molecular específica del neuroblastoma y de utilidad en el diagnóstico diferencial con otros tumores de células pequeñas redondas (17).
Rabdomiosarcoma
Corresponde a una neoplasia primitiva de células pequeñas redondas, con diferenciación músculo esquelética. En la infancia se distinguen dos subtipos mayores: el embrionario (65% de los casos) y el alveolar (20% de los casos), siendo este último de peor pronóstico. Ambos expresan miogenina y Myo-D1, que son genes “master reguladores” responsables de la diferenciación muscular (18). Otros marcadores inmunohistoquímicos útiles son la desmina y la actina sarcomérica, entre otros. La microscopía electrónica aporta hallazgos patognomónicos al identificar estructuras relacionadas con el sarcómero, como miofilamentos y bandas Z (12).
El rabdomiosarcoma alveolar presenta una translocación característica t(2;13) (q35;q14) en 75% de los casos, con fusión del gen PAX3 y FKHR (19). Una translocación asociada con menor frecuencia (10% de los casos) es la t(1;13)(p36;q14), con fusión de los genes PAX7 y FKHR. Estudios han demostrado que estas translocaciones tienen también un valor pronóstico, siendo la fusión PAX3-FKHR un factor de pronóstico adverso, mientras que la fusión PAX7-FKHR tendría una evolución más favorable (20). Además, en pacientes con enfermedad metastásica existe una importante diferencia en el pronóstico, con una sobrevida a 4 años de 75% para PAX7-FKHR versus 8% para PAX3-FKHR (21).
Para el rabdomiosarcoma embrionario no se han comunicado cambios genéticos tumor específicos.
Linfoma
Los linfomas que entran en el diagnóstico diferencial de los tumores de células pequeñas redondas de la infancia son el linfoma linfoblástico y el linfoma de Burkitt. Aquí, el panel inmunohistoquímico es de gran utilidad. El linfoma linfoblástico es TdT positivo y expresa marcadores de células T en 80 a 85% de los casos (CD3, CD43) y en 15 a 20% muestra marcadores de células B, como el CD20. El linfoma de Burkitt en su totalidad es de estirpe B, expresa CD19, CD20 y CD10 en algunos casos, con negatividad para el TdT.
Las neoplasias hematológicas poseen translocaciones que comprometen rearreglos de genes de inmunoglobulinas (linfomas de células B) o de genes de los receptores de células T (linfomas de células T), los que no están presentes en neoplasias no linfoides. En el linfoma de Burkitt se observa la translocación característica t(8;14) (q23;21) en 80% de los casos y también se puede detectar el genoma del EBV en la forma endémica. El linfoma linfoblástico muestra las translocaciones t7q34, t14q11 y t7p15 en un tercio de los casos (1).
Tumor desmoplástico de células pequeñas redondas
Es una neoplasia rara, muy agresiva, que ostenta el peor pronóstico dentro de los tumores de células pequeñas redondas. Las técnicas de inmunohistoquímica muestran expresión simultánea de marcadores epiteliales (queratina), musculares (desmina) y neurales (enolasa neuro-específica). El hallazgo citogenético característico y diagnóstico es la presencia de la translocación t(11;22)(p13;q12) (Fig. 5), con fusión de los genes EWS y WT1 (22 y 23) (Fig. 6).
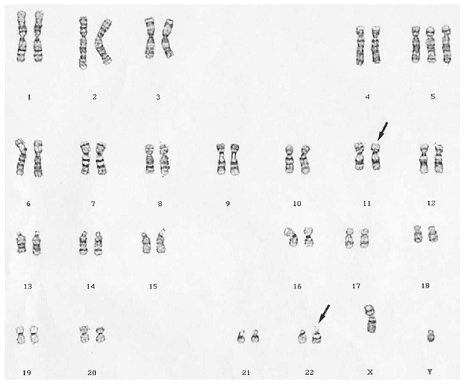 Tamaño completo
Tamaño completo Figura 5. Cariotipo de un paciente con tumor desmoplástico de células pequeñas redondas (DSRCT). Las flechas indican los cromosomas comprometidos en la t(11;22) (p13;q12) característica
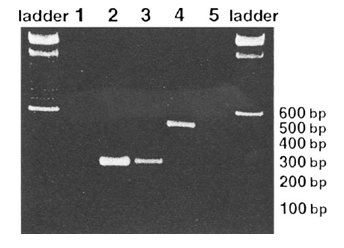 Tamaño completo
Tamaño completo Figura 6. Análisis RT-PCR de RNA tumoral para transcriptos quiméricos EWS-WT1. El panel representa el gel de agarosa en el cual se realizó la electroforesis de los productos de PCR. Los tamaños de los fragmentos se muestran a la derecha. Los carriles 2, 3 y 4 representan tres diferentes casos de DSRCT y el carril 5 es RNA de músculo esquelético normal. Los productos de 270 pares de bases (carriles 2 y 3) son los tamaños más comúnmente detectados en transcriptos de RNA quiméricos EWS-WT1. El transcripto en el carril 4 indica un corte alternativo que puede ocurrir tanto en el gen EWS como en el WT1
Osteosarcoma de células pequeñas
Corresponde a una variante rara, que representa 1 a 2% de todos los osteosarcomas. Morfológicamente puede simular un tumor de Ewing o un linfoma, sin embargo, a diferencia de éstos muestra variable formación de osteoide, que si es focal puede dificultar su diagnóstico. No presenta marcadores inmunohistoquímicos propios ni anormalidades cromosómicas características (5).
Condrosarcoma mesenquimal
Variante de condrosarcoma (2% del total), caracterizada por un patrón bifásico, en el que islas de cartílago bien diferenciado aparecen junto con un componente indiferenciado de células pequeñas redondas y azules, que eventualmente puede constituir un problema diagnóstico, debido al incremento de muestras obtenidas por punción con aguja fina.
Un estudio analizó la utilidad de la identificación inmunohistoquímica del producto del gen Sox9 para distinguir al condrosarcoma mesenquimal de otras neoplasias de células pequeñas redondas, siendo este marcador fuertemente expresado sólo por el condrosarcoma mesenquimal (18). Sox9 forma parte de un grupo de genes “master reguladores” que controlan distintas vías de diferenciación mesenquimática y que se expresan durante la embriogénesis; se ha demostrado que su transcripción juega un rol en la diferenciación temprana de condrocitos (24). No se han encontrado hallazgos citogenéticos consistentes (25).
Conclusiones
La contribución de la genética molecular ha mejorado significativamente la precisión en el diagnóstico de los tumores pediátricos de células pequeñas redondas; sin embargo, no se debe olvidar que el primer enfoque siempre debe ser la evaluación morfológica convencional, suplementada, en caso necesario, con técnicas auxiliares como la inmunohistoquímica, como una forma de dirigir mejor el diagnóstico. Sólo en la minoría de los casos, cuando los resultados sean no concluyentes o discordantes, será necesario recurrir al estudio genético molecular. Por otro lado, no se debe perder de vista que, si bien la especificidad de la fusión de genes es razonablemente alta, no es absoluta.
La detección de translocaciones características no sólo tiene importancia diagnóstica, sino que varios estudios han asociado su presencia y la de otras alteraciones genéticas con un valor pronóstico; además, el conocimiento de la patología molecular de estos tumores podría también servir para desarrollar terapias blanco. Actualmente, el uso de la tecnología de los microarrays ha permitido estudiar los perfiles de expresión genética de diversos tumores y descubrir genes cuya expresión es de utilidad en el diagnóstico diferencial de las neoplasias de células pequeñas redondas, como el neuroblastoma; o, a través del análisis de cluster, predecir pronóstico y asignar a los pacientes a grupos de riesgo determinados.
Referencias
- Tarkkanen M, Knuutila S. The diagnostic use of cytogenetic and molecular genetic techniques in the assessment of small round cell tumours. Current Diagnostic Pathology 8: 338-348, 2002.
- Chang Ch, Shidham V. Molecular genetics of pediatric soft tissue tumors. Journal of Molecular Diagnostics 5: 143-154, 2003.
- Dehner L. Primitive neuroectodermal tumour and Ewing´s sarcoma. Am J Surg Pathol 17: 1-13, 1993.
- Khoury J. Ewing Sarcoma Family of Tumors. Adv Anat Pathol 12: 212-220, 2005
- de Alava E. Diagnosis of small round cell tumours of bone. Current Diagnostic Pathology 7: 251-261, 2001.
- Zoubeck A, Pfleiderer C, Sazer-Kuntschik M, et al. Variability of EWS chimaeric transcripts in Ewing tumors: a comparision of clinical and molecular data. Br J Cancer 70: 908-913, 1994.
- Zucman J, Melot T, Desmaze C, et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumors. EMBO J 12: 4481-4487, 1993.
- Jeon I-S, Davis N, Braun B S, et al. A variant Ewing´s sarcoma translocation t(7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 10: 1229-1234, 1995.
- Kaneko Y, Yoshida K, Handa M, et al. Fusion of an ETS-family gene E1AF, to EWS by t(17;22) (q12;q12) chromosome translocation in an undifferentiated sarcoma of infancy. Genes Chromosomes Cancer 15: 115-121, 1996.
- Peter M, Couturier J, Pacquement H, et al. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 14: 1159-1164, 1997.
- Zoubeck A, Dockhorn-Dworniczak B, Delattre O, et al. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol 14: 1245-1251, 1996.
- Mierau G, et al. Role of electron microscopy and other techniques in the diagnosis of childhood round cell tumors. Hum Pathol 29: 1347-1355, 1998.
- White P S, Maris J M, Beltinger C, et al. A region of consistent deletion in neuroblastoma maps within human chromosome 1p36.2-36.3. Proc Natl Acad Sci USA 92: 5520-5524, 1995.
- Schwab M, Alitalo K, Klempnauer K-H, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumor. Nature 305: 245-248, 1983.
- Brodeur G M, Nakagawara A. Molecular basis of clinical heterogeneity in neuroblastoma. Am J Pediatr Hematol Oncol 14: 111-116, 1992.
- Bown N, Cotterll S, Latowka M, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. New Engl J Med 340: 1954-1961, 1999.
- Gilbert J, Haber M, Bordow S, Marshall G, Norris M. Use of tumor-specific gene expression for the differential diagnosis of neuroblastoma from other pediatric small round cell malignancies. Am J Pathol 155: 17-21, 1999.
- Wehrli B M, Huang W, Ayala A, Czerniak B, et al. Sox9, a Master Regulator of chondrogenesis, distinguishes mesenchymal chodrosarcoma from other small blue round cell tumors. Hum Pathol 34: 263-269, 2003.
- Ladanyi M. The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 4: 162-173, 1995.
- Anderson J, Gordon T, McManus A, Mapp T, Gould S, Kelsey A, et al. Detection of PAX3-FKHR fusion gene in paediatric rhabdomyosarcoma: a reproducible predictor of outcome? Br J Cancer 85: 831-835, 2001.
- Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostics indicators in alveolar rhabdomyosarcoma: a report from the children´s oncology group. J Clin Oncol 20: 2672-2679, 2002.
- Lae M, Roche P, Jin L, Lloyd R, Nascimiento A. Desmoplastic small round cell tumor: a clinicopahologic, inmunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol 26: 823-835, 2002.
- Sandberg A, Bridge J. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: desmoplastic small round-cell tumors. Cancer Genetics and Cytogenetics 138: 1-10, 2002.
- Huang W, Zhou X, Lefebvre V, et al. Phosphorylation of Sox9 by cyclic AMP-dependent protein kinase A enhances Sox9´s ability to transactivate a Col2a1 chondrocyte-specific enhancer. Mol Cell Biol 20: 4149-4158, 2000.
- Nakashima Y, Park Y-K. Mesenchymal chondrosarcoma. In: Fletcher C D (eds) Pathology and Genetics of Tumours of Soft Tissue and Bone. In: Kleihues P (eds) WHO Classification of Tumors. IARC, 2002, in press.
