Case reports
← vista completaPublished on August 23, 2024 | http://doi.org/ 10.5867/medwave.2024.07.2789
Ectopia cordis: reporte de un caso
Thoracic ectopia cordis: A case report
Abstract
Ectopia cordis is a congenital heart malformation of the sternal wall, with a prevalence of 0.1% among heart conditions and an incidence of 5.5 to 7.9 per million births. It is characterized by the heart being located outside the thoracic cavity, and it may be accompanied by other congenital anomalies such as omphalocele, Cantrell´s pentalogy, or Fallot´s tetralogy. We present a case of thoracic ectopia cordis in a male neonate. After birth, we also observed a midline thoracic malformation and respiratory difficulties with clinical and paraclinical features consistent with tetralogy of Fallot. It was decided to provide skin flap coverage, and due to the poor prognosis of the heart condition, palliative care was chosen. Unfortunately, the neonate passed away after seven days. This clinical case study contributes to understanding this rare condition and may help improve diagnosis and treatment of affected patients.
Introduction
Ectopia cordis is a congenital heart malformation affecting the sternal wall, with a prevalence of 0.1%, affecting approximately 5.5 to 7.9 per million births. The abnormal position of the heart outside the thoracic cavity characterizes the newborn. Additionally, the patient may present other congenital anomalies such as omphalocele, Cantrell´s pentalogy, or Fallot´s tetralogy [1]. Ectopia cordis is often associated with high morbidity and mortality rates due to both cardiac and extracardiac defects [2]. Caring for the mother-infant pair becomes a public health priority. In primary care, it is essential to establish early diagnosis of these patients to provide immediate and efficient care to the newborn [3].
The etiology of ectopia cordis remains unknown. Some hypotheses suggest that the heart does not descend properly during fetal development, leading to an abnormal midline formation. The heart does not descend properly, leading to an abnormal midline formation. Other theories propose the rupture of the yolk sac and/or chorion, affecting midline formation due to fibrous tissue preventing normal fusion under increased pressure. Another hypothesis mentions the absence of a protein gene, which plays a role in the embryonic primary heart development during the fourth week when cardiac positioning typically begins in the normal definitive location, yet the body wall fails to close [4,5,6].
Advancements in technology have allowed the identification of embryonic malformations like ectopia cordis as early as the first trimester of pregnancy, with up to 90% efficacy in prenatal diagnosis. Efficient diagnostic tools, including prenatal ultrasound, postnatal echocardiography, and magnetic resonance imaging, can effectively diagnose complex malformations and multiple abnormalities. Early diagnosis, which facilitates immediate treatment of newborns, is crucial to increase their chances of survival [7,8,9].
The care and treatment of patients with ectopia cordis requires a multidisciplinary team consisting of professionals in neonatology, nursing, radiology, perinatology, pediatric cardiology, and pediatric surgery to repair diaphragmatic, sternal, pericardial, intracardiac, or omphalocele defects. In this regard, attempting to reposition the heart within the thoracic cavity is necessary. The most common complication of this process is the increase of intrathoracic pressure, leading to vascular compression and decreased tissue perfusion [10].
Depending on the severity of the defect, ectopia cordis can be classified as a) partial, characterized by a sternal fissure ranging from a "V" shaped interruption to a complete division of the sternum, allowing the heart to be seen beating beneath a thin transparent layer, and b) complete, as in the case presented in this article, involving the complete exposure of the heart outside the thoracic cavity without pericardial coverage; this type of defect is extremely rare [11].
In neonates with ectopia cordis, it is essential to rule out other defects, as it is common to find syndromes such as PHACES, Beckwith-Wiedemann, Proteus, Klippel-Trenaunay, Russell-Silver, and type 1 neurofibromatosis. This can be achieved with X-rays, ultrasounds, and computed tomography to provide a holistic view of the patient’s anatomy, physiology, and signs for proper and efficient care [11].
Another classification of ectopia cordis is based on the heart's location: a) cervical, b) cervicothoracic, c) thoracic, and d) thoracoabdominal or abdominal. In this context, the ectopic heart may be entirely exposed, covered by skin, or enclosed by a pericardium serous. Due to the rarity of ectopia cordis, there are no established guidelines to facilitate accurate diagnosis and proper management of this potentially life-threatening malformation [12].
In congenital pathologies such as ectopia cordis, prenatal diagnosis makes a significant difference in the patient's chances of survival. Previous case studies have shown the difficulties in identifying these malformations, as they depend on the constant monitoring of the pregnancy, complex diagnostic studies, and the expertise of specialists to visualize the defects. Mukherjee [13], explains in a case report that a pregnant woman underwent ultrasounds during the first and second trimesters without detecting ectopia cordis; however, an ultrasound at 32 weeks of gestation revealed the pathology. This suggests that defects become more visible as the fetus advances in gestational age. Establishing the type of malformation before birth can lead to a better-equipped environment for delivery, potentially improving the prognosis, neonatal resuscitation, and patient stabilization [14,15].
Case report
We obtained informed consent from the parents and institutional authorization from a Mexican public hospital to publish this report. A male neonate patient weighing 3.3 kg, measuring 50 cm in height, and with a body surface area of 0.22 m2, as seen in Figure 1. He was born by vaginal delivery at term, with a limited number of prenatal visits and no reported preexisting maternal illnesses or drug use.
Ectopia cordis thoracis.
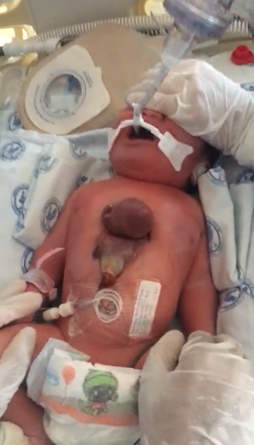
Source: Provided by the authors.
After birth, a midline thoracic malformation, respiratory distress, and protrusion of the heart outside the thoracic cavity were observed. The attending physician decided to perform endotracheal intubation and refer the patient to a tertiary-level hospital to manage these malformations.
An echocardiographic study was performed, determining the diagnosis of long-segment pulmonary atresia with ventricular septal defect due to misalignment of the conal septum, positioned anteriorly and to the left, without being able to adequately assess the source of the pulmonary flow. Computed tomography angiography of the heart and great vessels was conducted to complement the study.
The cardiac computed tomography angiography was carried out using a 128-detector scanner, employing a non-electrocardiogram-synchronized angiographic protocol. A total of 5 mL of iodinated contrast medium at 300 mg/mL was administered at 1.5 ml/kg using the 'bolus tracking visual' technique. The average heart rate during the study was 110 beats per minute.
The findings reported in the imaging tests were as follows:
-
Situs solitus of abdominal, bronchial, and atrial structures, meaning the typical location of these structures with respect to the midline.
-
Ectopia cordis.
-
Atrioventricular connection: Concordant with perforated mode.
-
Ventriculoarterial connection: Concordant with over-riding aorta (50% overriding).
-
Systemic venous returns: A double system of the superior vena cava is observed. The right superior vena cava connects to the right atrium without obstructions. The persistent left superior vena cava drains into the coronary sinus. The retrohepatic portion of the inferior vena cava connects to the right atrium without obstructions.
-
Normal pulmonary venous returns.
-
Pulmonary trunk: 1.6 mm.
-
Right pulmonary branch: 1.3 x 2 mm.
-
Left pulmonary branch: 1.5 mm.
Furthermore, the following associated lesions were observed:
-
Ventricular septal defect due to anterior displacement of the conal septum, measuring 8mm x 6mm.
-
Atrial septal defect (ostium secundum) of 4 mm.
-
Left aortic arch, supra-aortic vessels with normal origin, and at least three aortopulmonary collaterals to the left lung were identified, along with one more originating from the transverse portion of the aortic arch to the right lung.
-
Severely hypoplastic infundibulum. No pulmonary valve plane was identified.
-
Atelectasis in the right upper pulmonary lobe.
The studies determined a diagnosis of ectopia cordis combined with pulmonary atresia (infundibulum, valve, and pulmonary trunk), ventricular septal defect, pulmonary artery hypoplasia, and non-communicating aortopulmonary collaterals (Barbero-Marcial type B). In this context, the therapeutic decision was to perform closure or coverage of the defect with a skin flap, as seen in Figure 2. However, the patient was under permanent monitoring by the cardiology department due to the poor prognosis of his heart condition. The patient was admitted to palliative care with parental consent, and after seven days, he passed away.
Extra thoracic lined neonatal heart.
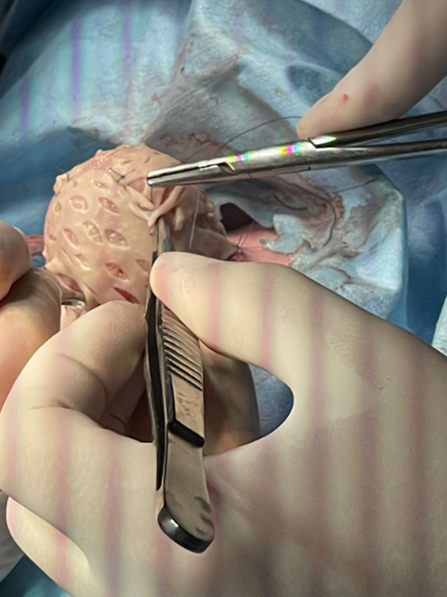
Source: Provided by the authors.
Discussion
The importance of early diagnosis and treatment of ectopia cordis is crucial to improve the prognosis and even the survival of a neonate with this condition. As with any fetal anomaly, a thorough investigation of all fetal systems should be carefully examined. Treatment options for ectopia cordis generally require a multi-surgical approach. Repair of the omphalocele, sternal, diaphragmatic, and pericardial defects is included in the initial phase, followed by soft tissue coverage of the heart.
Soft tissue coverage protects the heart and prevents fluid loss and cardiac desiccation. The repair of intracardiac anomalies and the reduction of the heart into the thoracic cavity are typically performed in the second phase; surgical correction can be challenging due to thoracic cavity hypoplasia [16]. In the case described, the first part of the treatment was successfully completed; unfortunately, the patient passed away before a second surgical procedure for complete cardiac reduction could be performed.
Complete repair of ectopia cordis is unlikely due to multiple complications in patients; there are few long-term survivors even after repair [17]. In the presented case, the patient had a complete defect with multiple comorbidities, had a reserved prognosis, and was referred to palliative care, understanding that survival was unlikely due to the complexity of his condition.
When dealing with congenital anatomical defects in the thoracic wall, it is recommended to have an antepartum diagnosis, preferably during the first or second trimester of gestation, through ultrasound and magnetic resonance imaging. In this way, arranging a multidisciplinary group to care for the newborn will be decisive in the prognosis and treatment. Kalaniti [18] recorded a case where the patient was born vigorous but died during transport to a third-level institution due to cyanosis and desaturation. In the present clinical case, the patient was born by vaginal delivery, and only after birth was the malformation observed and required intubation; he was studied with ultrasound and angiotomography and was successfully transferred to the tertiary level. It was evident that he had no previous diagnosis, and undergoing the whole labor complicated his health situation due to the malformation conditions. Torres et al. [19] point out that in cases of ectopia cordis, the prenatal imaging characteristics can be complex and challenging and often require the radiologist to have a high level of suspicion and familiarity with the imaging patterns.
Among the common comorbidities associated with ectopia cordis are ventricular septal defect, atrial septal defect, pulmonary stenosis, right ventricular diverticulum, double right ventricular outflow tract, tetralogy of Fallot, omphalocele, gastroschisis, cleft lip and palate, scoliosis, and central nervous system malformations [20]. In another case report, prenatal ultrasound detected ectopia cordis associated with a complex intracardiac defect, which included a common atrium communicated with a single ventricle through a common atrioventricular valve. Additionally, multiple extracardiac defects were observed, including skeletal and cranial abnormalities. The skeletal anomalies included a dysplastic right upper limb, short bones with satisfactory calcification, and constrictive anomalies in the left upper limb finger. Brain tissue was partially herniated, and the parents opted for therapeutic abortion.
The situation of a neonate with a malformation, as well as syndromes that affect the patient´s quality of life and their families, has allowed some countries to legally terminate the pregnancy and proceed with a fetal autopsy to determine the cause or damage [21]. Among other studies, chorionic villus sampling is performed in advance to assess fetal karyotype and store deoxyribonucleic acid further to determine a higher risk of recurrence. It can be argued that one advantage of diagnosing fetal anomalies during the first trimester is the ability to perform surgical termination if suitable for the parents. In the described case, the malformation was not determined beforehand, and the parents had no alternative. Unfortunately, with the neonate´s passing, no tests were conducted [22].
One case of ectopia cordis generated significant interest in the scientific community as a patient with a spontaneous dichorionic diamniotic twin pregnancy was identified via ultrasound. Twin A had an isolated complete ectopia cordis with no other morphological malformation, while Twin B had no abnormalities. The parents chose not to undergo selective feticide and opted for postnatal palliative care and cardiac covering. The newborn passed away within a few hours of the cesarean section [23].
In other cases, treatment and favorable outcomes in intrauterine-diagnosed neonates were described. In one case, a multidisciplinary team was assembled to plan the treatment of ectopia cordis. A full-sized 3D printed model created from a magnetic resonance imaging scan was used for stabilization, wrapping the heart in sterile plastic with a suture at the cardiac apex [24]. The heart was reduced into the left thorax with partial sternal closure and skin flap mobilization. Subtotal diaphragmatic reappraisal was performed with intentionally delayed reduction of omphalocele, allowing more space for the cardiac cavity in the body. Bandages were applied to the omphalocele twice daily until epithelialization. The total hospitalization lasted 4.5 months, and survival of 19 months was reported at the time of publication.
Conclusions
Early diagnosis and treatment of ectopia cordis are important and determine the prognosis of the newborn, considering that the prognosis is reserved due to the complicated nature of this malformation. It has been found that early diagnosis and timely treatment for both the mother and the newborn improve survival and reduce comorbidities when appropriate diagnostic resources, specialized intensive care, and a multidisciplinary healthcare team are available.
Fetuses or neonates diagnosed with ectopia cordis should undergo a comprehensive multisystem analysis because they often present multi-organ damage and, at times, associated syndromes. Gestational age and birth weight are intrinsic variables, while extrinsic variables include preparation for receiving the newborn and optimal conditions for resuscitation and hemodynamic stabilization.
Neonates with ectopia cordis face a daunting treatment journey characterized by its complexity and high level of risk. The condition demands meticulous surgical interventions aimed at reducing the cardiac cavity, typically performed in multiple stages. Initially, the exposed heart must be carefully covered to shield it from potential damage and minimize the risk of infection. Subsequent procedures focus on thoracic reduction and correction, demanding a high degree of surgical expertise and precision due to the delicate nature of the involved cardiac and thoracic structures.
The decision-making process for parents of fetuses diagnosed with ectopia cordis prenatally is fraught with challenges. Factors such as the severity of the condition, the presence of other significant deformities, and legal considerations regarding the termination of pregnancy all weigh heavily on their minds. While some parents may opt for termination upon early diagnosis, especially when faced with a poor prognosis or significant associated anomalies, others choose to continue the pregnancy.
For those who proceed with the pregnancy, despite the challenges posed by ectopia cordis, palliative care may be provided to manage symptoms and ensure the baby´s comfort until their passing. This compassionate approach acknowledges the limitations of available interventions and prioritizes the infant´s quality of life. However, parental decisions regarding the continuation or termination of the pregnancy are deeply personal and influenced by cultural, religious, and ethical beliefs, underscoring the importance of individualized care.
Despite the grim prognosis associated with ectopia cordis in some cases, there are instances where surgical correction offers hope for improved outcomes and long-term survival. These cases highlight the importance of ongoing advancements in pediatric cardiac care and the need for tailored treatment approaches that consider each family's unique circumstances and preferences.
