Clinical reviews
← vista completaPublished on October 22, 2018 | http://doi.org/10.5867/medwave.2018.06.7298
Current knowledge on Hashimoto's encephalopathy: a literature review
Conocimientos actuales en encefalopatía de Hashimoto: revisión de la literatura
Abstract
Hashimoto's encephalopathy is a rare disease, with a reported prevalence of 2.1 per 100 000. Clinical manifestations include confusion, decreased state of consciousness, cognitive deficit, seizures, myoclonus, ataxia, and focal neurological deficits. Due to the wide variety of signs and symptoms, clinical diagnostic suspicion is essential. Diagnosis is based on three pillars: the presence of neurological clinical manifestations after ruling out other causes of encephalopathy. 2) Presence of increased antithyroid antibodies. 3) Significant clinical improvement after the administration of immunomodulation. The treatment of Hashimoto's encephalopathy pursues two objectives: to control the autoimmune process and to control the complications of the disease. Although in most cases recovery is complete with treatment, the risk of relapse can range from 12.5 to 40% in follow-ups to 2 years.
|
Key ideas
|
Introduction
Hashimoto’s encephalopathy is a disease that was originally reported by Brain et al. in 1966[1], who reported a possible autoimmune mechanism of action[2]. It is a rare disease characterized by the presentation of encephalopathy with diverse neuropsychiatric manifestations, and positive antithyroid titers[3]. It should be considered as a differential diagnosis in patients without history of neurological disease[4] and after ruling out other causes such as infection[5]. In most cases, thyroid function tests are normal, and corticosteroids elicit a rapid remission of the clinical manifestations[6].
Due to its unusual presentation, it is important to be aware of the clinical characteristics associated with this pathology for a timely and precise diagnosis and treatment, because if left untreated, this condition may be potentially fatal.
The objective of this review is to provide a glimpse of the current knowledge on the pathophysiology, diagnosis, and management for the specialist.
Methods
This article is based on the systematic review of various databases and a critical analysis of the literature. Due to the rarity of this condition, the available evidence is mainly based on observational studies; therefore, the articles included in our review are mostly case series and case reports, as well as narrative reviews.
Inclusion criteria
In a first instance, all the retrieved publications in Spanish and English in the last five years were included, and any relevant references published before the search timeframe that were important for the discussion were also included. Cases described only as abstracts at meetings and/or letters to the editor were excluded because of conciseness. In a second instance, each case report or case series was evaluated individually and those where specific values of antithyroid antibodies were not considered (phrases such as "high antibodies" or "antibodies outside the normal range") or where the reported value of the antibody titers was not high enough to be considered diagnostic (that is, at least one antithyroid antibody titer that exceeded the minimum cutoff point of 100 µUI/mL) were excluded (see Figure 1).
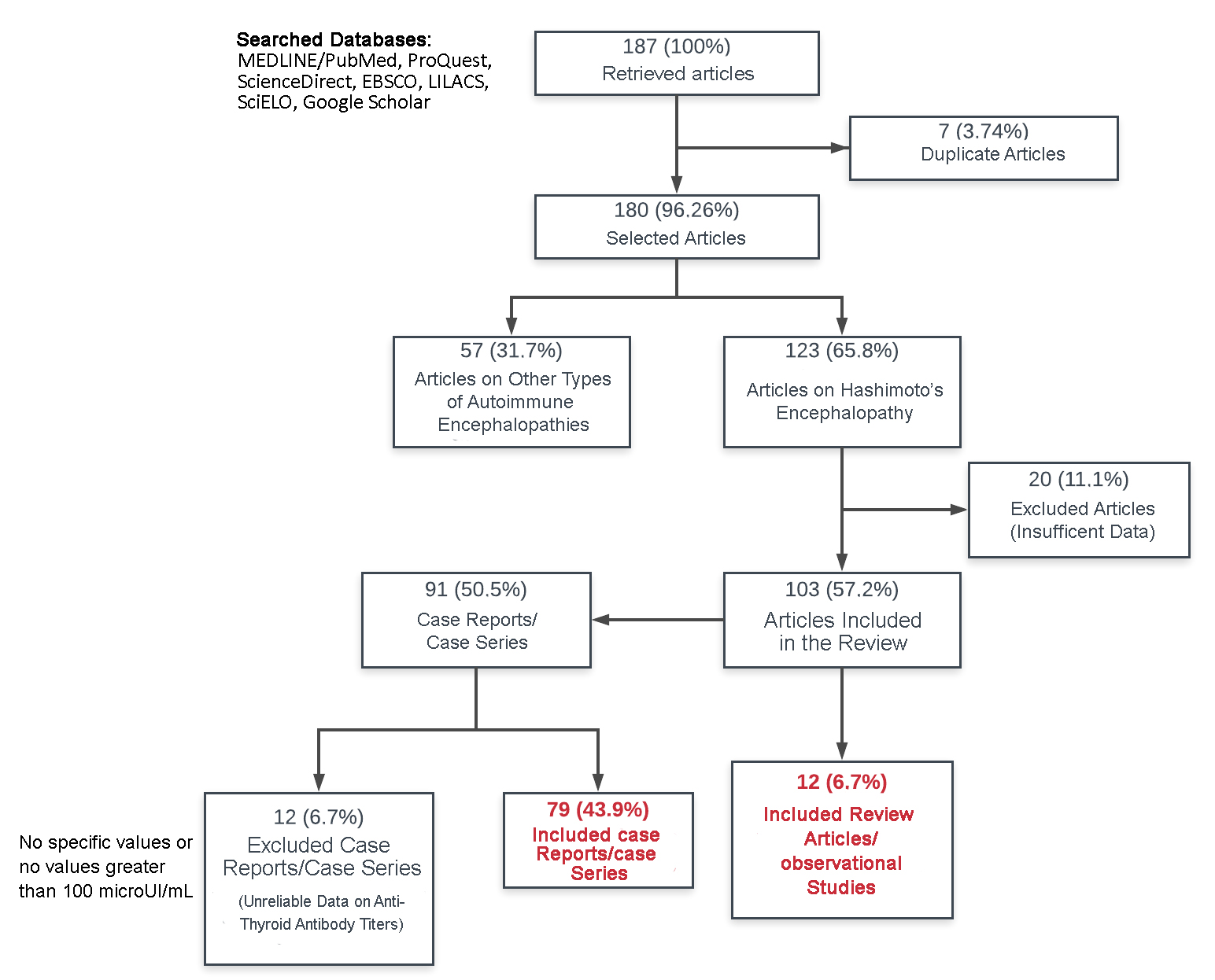 Full size
Full size Search strategy
Publications available in main databases such as MEDLINE/PUBMED, ScienceDirect, ProQuest, EBSCO, LILACS, SciELO, and other search engines such as Google Scholar were evaluated. The key terms, “Hashimoto’s encephalopathy”, “steroid-responsive encephalopathy associated with autoimmune thyroiditis” and “Hashimoto's encephalitis” were searched in English and Spanish within title and abstract domains, using Boolean operators “AND” and “OR”. This process was carried out independently by both investigators.
Results
Prevalence
Hashimoto’s encephalopathy is a rare disease. With a reported prevalence of 2.1 cases/100,000 population, it predominantly affects adults starting at 50 years old, and females (female-to-male ratio of 5:1)[7] In children, there are reports of presentation starting from 14 months of age[8], although it most frequently affects adolescents[9] with a peak at 14 years old.
Causes
Hashimoto’s encephalopathy has a presumable autoimmune etiology. Its clinical presentation, as well as its predominance in females and its coexistence with other autoimmune diseases such as systemic lupus erythematosus, myasthenia gravis, and other collagen disorders in up to 30% of the cases[10] place this pathology within the group of immunologic diseases.
Due to its autoimmune nature, recently some authors have renamed it “encephalopathy associated with autoimmune thyroiditis”[11]. This name hints better at its etiology and gives us the option to associate it to the whole range of antibodies found in thyroid autoimmune conditions, not just those closely related to Hashimoto's thyroiditis (antibodies against thyroid peroxidase, TPO). The decision to retain the traditional name of Hashimoto's encephalopathy in this review is intended to facilitate the bibliographic search.
Regarding the pathophysiology, there are currently two possible explanations. The first, the most accepted, states that anti-thyroglobulin (anti-Tg), anti-thyroid peroxidase (anti-TPO), or anti-TSH receptor (anti-TSHR, also known as thyrotropin receptor antibodies, TRAb) antibodies would trigger the pathophysiological cascade in this disease. There is supporting evidence that anti-TPO antibodies bind to astrocytes and alter central nervous system function[3].
The second explanation refers to other antibodies directed against autoantigens of the cerebral vascular endothelium causing damage to neuronal tissues[12]. High titers of anti-α-enolase –a 34-kDa cytosolic protein of the erythrocyte glycolytic pathway[13] common for thyroid and cerebral tissue[5]– were found in patients with Hashimoto's encephalopathy who had excellent responses to corticoids[14]. These antibodies would attack only the terminal N-amino end of the α-enolase[15] and could be used as a confirmatory diagnostic test[16].
Other antibodies currently under investigation are IgG antibodies against dimethylargininase I (a 36-kDa protein present in neuronal and endothelial cells) and aldehyde reductase I (present in vascular and neuronal cells)[11],[17]. Less clearly, antibodies against parietal cells or intrinsic factor have also been implicated[18]. The elucidation of the pathophysiology is far from complete.
The literature proposes two forms of presentation. The first is a recurrent vasculitic form with cerebral edema and a reduced cerebral blood flow due to microvascular damage in an insidious manner, presenting with cognitive decline and stroke-like features. A second progressive form is expressed as an encephalopathy (confusion, psychosis, coma), where antithyroid antibodies would cross the blood-brain barrier producing brain injury[19].
Clinical manifestations
Although classic Hashimoto´s encephalopathy has been described as a neuropsychiatric syndrome that causes unresponsiveness in varying degrees of severity, causing even coma[20], its spectrum of neurological manifestations is quite broad and may predate established encephalopathy by days to months. Therefore, it is possible to distinguish 2 distinct stages:
Pre-encephalopathy: Neurological manifestations in this stage are diverse and can simulate almost any central nervous system pathology. In a case series of 13 patients, the most frequently observed manifestations were cognitive impairment and behavioral changes in 10 patients (predominantly with frontal and temporal involvement in neuropsychological evaluation), seizures in 6, sleep disorders in 9 (hypersomnia in 6 and hyposomnia in 3), and headache in 4 patients4. It has also been associated with sudden-onset narcolepsy with cataplexy[21].
There are much more complex reported cases where the predominant neurological alterations are psychiatric features such as catatonia[22], auditory hallucinations, psychosis[23], persecutory delusions, and mood disorders, sometimes misdiagnosed as senile dementia[24] or masked by depression in elderly subjects[25]. Not all patients present this stage.
Encephalopathy: Sudden or progressive unresponsiveness that can be isolated or preceded by a pre-encephalopathic stage. In both cases, the key for the diagnosis is the presence of elevated antithyroid antibody titers and a rapid clinical improvement after starting immunomodulating therapy[26].
Additionally, for didactic purposes, we can also classify Hashimoto´s encephalopathy according to the central nervous system manifestation that predominates in the patient, whether in the pre-encephalopathic or the encephalopathic stage (see Table 1).
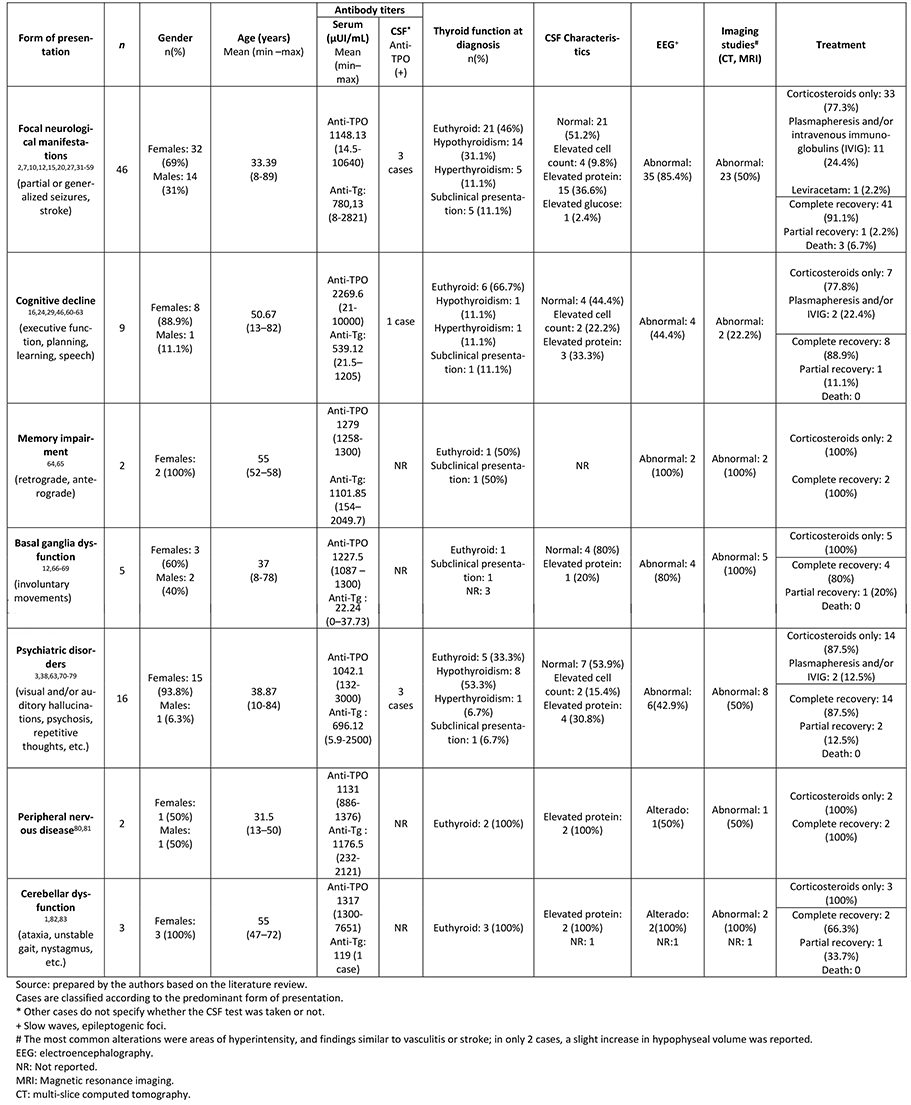 Full size
Full size Autonomic disturbances such as hypotension, bradycardia, and hypothermia are related to hypothalamic and neurovegetative ganglion involvement[2] and, in these cases, it is important to rule out involvement of the hypothalamic–pituitary–adrenal axis. Cerebellar involvement associated or not with encephalopathy may be manifested as hypotonia, opsoclonus, dysmetria, and dysdiadochokinesia[1]. In both cases, the form of presentation is a manifestation of a generalized cerebral vasculitis.
The duration of the clinical manifestations is variable. Durations between two days up to 8 months have been reported[18], depending on the time of initiation of treatment, although the most frequent presentation is a sudden onset with a rapid clinical improvement after the onset of corticosteroid therapy.
The presence of thyroid function abnormalities can lead to confusion at the time of diagnosis. Uncompensated hypothyroidism may present neurological complications such as seizures, dementia, or psychosis, but it differs from Hashimoto's encephalopathy in that the neurological manifestations improve after thyroxine replacement. Less frequently, it is associated with hyperthyroidism[27], which requires ruling out thyrotoxic psychosis. In any case, clinical suspicion leads to the diagnosis.
Some metabolic disorders such as hyponatremia, hyperglycemia, or uremia may coexist with the presentation of encephalopathy[28], which makes a correct diagnosis more difficult to achieve. In any case, unsuccessful management of the specific metabolic disturbances may lead to the differential diagnosis.
Diagnosis
The diagnosis of Hashimoto´s encephalopathy is an exclusion diagnosis; it is necessary to first rule out all possible pathologies afflicting the central nervous system (tumoral, infectious, autoimmune, etc.), as well as metabolic pathologies and paraneoplastic syndromes.
Although there are no globally accepted diagnostic criteria for Hashimoto´s encephalopathy yet, its diagnosis is based on the presence of central nervous system clinical manifestations such as: confusion, decreased level of consciousness, cognitive decline, seizures, myoclonus, ataxia and/or focal neurological deficits[29],, with exclusion of other possible causes of encephalopathy[9].
In addition, it is essential to have evidence of significantly elevated titers for at least one antithyroid antibody. The literature indicates that anti-TPO titers would have to be greater than 200 µUI/ml (more than 5 times the normal value) and some authors set a cut-off point of 500 µUI/ml as threshold[17]. Despite this, there are many published cases that do not follow this standard and make a diagnosis with much lower values (see Table 1), which casts doubt on the diagnosis. Given that a positivity to anti-thyroid antibodies is very prevalent in the healthy population and that the symptoms are diverse, the diagnosis of Hashimoto’s encephalopathy becomes difficult[30] and this is where questions arise: Is this disease a real condition? Are we just seeing the manifestations of a purely cerebral autoimmune pathology that we still don't know about?
We suggest using a cutoff value ≥ 100 µUI/mL of any antithyroid antibody to substantiate the diagnostic suspicion, but a higher cutoff value (≥ 200 µUI/ml) in at least one of the 3 types of antithyroid antibodies (anti thyroid peroxidase, anti-thyroglobulin and/or anti-TSH receptor) would make the diagnosis more evident. In any case, after ruling out other central nervous system pathologies, the diagnosis would be confirmed with a marked clinical improvement after corticotherapy.
Anti-thyroid antibodies are necessary but not sufficient for the diagnosis, and their titers are not directly correlated with the severity of the disease[84],[85]. Additionally, when the thyroid function is studied, it is found that most patients are euthyroid at the time of presentation of encephalopathy.
Cerebrospinal fluid analysis may show an inflammatory pattern with elevation of the protein level, whose values usually improve after treatment[84], although up to 80% of patients have normal characteristics[19]. Antithyroid antibodies may occasionally be isolated and their titers may be persistently elevated even after the patient's recovery[33]. It is important to notice that it is possible to find antithyroid antibodies in cerebrospinal fluid in patients with several thyroid diseases or other central nervous system pathologies such as neurosyphilis[86], without having evidence of Hashimoto’s encephalopathy at the time of testing.
The electroencephalogram usually renders nonspecific findings that may vary according to the specific neurological manifestations seen in patients. The most frequently observed abnormality was the presence of slow wave activity associated to encephalopathy[82] and in cases of seizures, epileptogenic foci could be observed.
Brain imaging study findings (computed tomography and/or magnetic resonance imaging) are in most of the cases normal or nonspecific, but these are crucial to rule out other etiologies. Between 49% and 50% of patients may present diffuse cerebral atrophy in cortical or subcortical locations[19], infarcts, and/or cortical focal abnormalities87. In 2 of the cases, abnormalities similar to lesions in the region of the sella turcica were reported.
Treatment
The treatment of Hashimoto’s encephalopathy has two objectives: the first, to halt the autoimmune process with the use of immunomodulators or plasma exchange, and the second, to control the complications of the disease with therapies such as antiepileptic drugs in case of seizures or mannitol in case of cerebral edema[38].
The first line of immunomodulating therapy are corticosteroids. Initial administration of intravenous methylprednisolone should be given for 3 to 7 days (adults: 1 g/day, children: 20–30 mg/kg/day)[33], followed by high doses of oral prednisone (doses of 1–2 mg/kg/day), which should be tapered down slowly according to clinical improvement[29],[88]. Maintenance doses can be administered for months up to 1-2 years[15].
Other immunomodulators include intravenous immunoglobulin, monoclonal antibodies, azathioprine, and methotrexate. Intravenous immunoglobulin is useful when patients have an incomplete response to corticosteroids[9] or when relapses occur during tapering down. In the case of monoclonal antibodies, the suggested dose of rituximab is 1,000 mg intravenously on days 1 and 14, and then once every 6 to 9 months depending on the white blood cell count[89]. Rituximab is well tolerated and induces a sustained remission without the need for additional corticosteroids[90].
Plasmapheresis was thought to work by removing circulating antithyroid antibodies, but as it has been shown that the severity of the disease is not related to the antibody titers, it is now thought that plasmapheresis may induce clinical improvement by removing other antibodies, autoimmune complexes, cytokines, and/or other inflammatory mediators currently unknown[62]. The number of plasma exchange courses may vary from 3 to 10[36].
Prognosis
Most patients improve completely after the onset of corticotherapy, but the risk of relapse may be as high as 12.5%-40% in a two-year follow-up[29], and 12.5% of patients do not respond to corticosteroids and will require immunosuppressors[84]. Frequency of presentation of neurological sequelae in adults has not been reported, but persistent neurological deficits were identified in more than 20% of children after a four-year follow-up[91].
Conclusion
The reviewed cases suggest that this disease is probably underdiagnosed, so it is reasonable to recommend that every patient with an inexplicable encephalopathy, especially women, is ruled out for Hashimoto’s encephalopathy. For this reason, it is necessary to request anti-thyroid antibodies.
Positivity for antithyroid antibodies, an electroencephalogram with slow waves and/or focal epileptiform activity that suggests the presence of epileptogenic foci, a brain cerebral magnetic resonance with normal findings or findings of vasculitis or diffuse ischemic lesions, and a clinical improvement after immunotherapy (corticosteroids, IVIG, or plasmapheresis) are helpful hints to make the diagnosis.
Notes
Author Contributions
IP: Concept and design, research, methodology, project management, monitoring, visualization, drafting of the manuscript, reviewing, and editing.
JP: Concept and design, research, methodology, monitoring, visualization, drafting of the manuscript, reviewing, and editing.
Acknowledgments
The authors appreciate the collaboration and excellent disposition of Dr. Jorge Benavides Vásquez, research scholar at Penn State College of Medicine (Hershey, PA, USA) in the translation of the original version from Spanish to English.
Conflict of Interest
The authors have completed the ICMJE's conflict of interest declaration form, and declare that they have not received funding for writing this article; that they have no financial relations with organizations that might have interest in the article in the last three years; and that they do not have any other relationships or activities that could influence the contents of the published article. The disclosure forms can be requested by contacting the author responsible or the editorial editor of the Journal.
Funding
The authors state that they had no external sources of funding.
From the editor
The authors originally submitted this article in Spanish and subsequently translated it into English. The Journal has not copyedited this version.
