Reporte de caso
← vista completaPublicado el 10 de enero de 2023 | http://doi.org/10.5867/medwave.2023.01.2634
Polirradiculoneuropatía desmielinizante inflamatoria crónica con anticuerpos antineurofascina-155: primer reporte de caso en Perú
Chronic inflammatory demyelinating polyradiculoneuropathy with antineurofascin-155 antibodies: A first case report in Peru
Abstract
Chronic inflammatory demyelinating polyradiculoneuropathy is a clinically heterogeneous group of immune- mediated peripheral neuropathies that share neurophysiological manifesta-tions of demyelination and albuminocytologic dissociation.
There are typical and atypical variants of this disease, some associated with antibodies against proteins of the node of Ranvier, such as neurofascin- 155.
We present the case of a 38- year- old male who presented with an eight- month history of par-esthesia and progressive weakness of four limbs associated with diplopia and dysphagia. The patient was conscious, with symmetric flaccid quadriparesis of distal predominance, hyp-otrophy in the dorsum and palm of both hands, generalized areflexia, postural low frequency, and high amplitude tremor in upper limbs of left predominance, appendicular dysmetria, dys-diadochokinesia, ophthalmoparesis to dextroversion in the right eye, absent gag reflex, ataxic gait with an increased base of support and positive Romberg's sign.
Cerebrospinal fluid showed albuminocytologic dissociation, and electromyography was com-patible with primarily demyelinating sensory- motor polyneuropathy.
Due to clinical suspicion, we requested anti- neurofascin- 155 antibodies, which tested positive.
The patient was treated with methylprednisolone at a dose of one gram per day for five days, followed by one milligram per kilogram for three months of prednisone, with progressive de-crease, which improved diplopia and dysphagia, with no effect on limb strength and even worsening of function. For this reason, treatment with rituximab was started in doses of two grams, presenting a substantial improvement in distal muscle strength, tremor, gait stability, coordination, and functionality measured with the modified Rankin scale.
Main messages
- Chronic inflammatory demyelinating polyradiculoneuropathy is a clinically heterogeneous entity with typical and atypical variants, some associated with antibodies against Ranvier’s node proteins such as neurofascin-155.
- We report a case of an atypical subtype of chronic inflammatory demyelinating polyradiculoneuropathy associated with anti-neurofascin-155 antibodies, which defines a specific clinical picture.
- Identifying antibodies in chronic inflammatory demyelinating polyradiculoneuropathy has therapeutic implications since they are markers of poor response to immunoglobulins and corticosteroids and excellent response to rituximab.
Introduction
Chronic inflammatory demyelinating polyradiculoneuropathy is an autoimmune disorder of the peripheral nervous system, representing the most common treatable chronic neuropathy in the world [1,2,3], with an estimated prevalence of three per 100,000 population [4]. This disease typically presents as a recurrent or slowly progressive neuropathy characterized by proximal and distal symmetric weakness developing over at least eight weeks [2,5]. Diagnosis is based on clinical and electrophysiological criteria, identifying patients who may respond to immunomodulatory therapy [6,7].
Antibodies directed against proteins in the Ranvier’s node have recently been described, defining atypical subtypes of chronic inflammatory demyelinating polyradiculoneuropathy with specific clinical features [1,8]. Anti-neurofascin-155 (anti-NF155) antibodies are the most frequently described in approximately 5 to 7% of patients with chronic inflammatory demyelinating polyradiculoneuropathy. They define a phenotype characterized by distal-dominant weakness, low-frequency and high-amplitude tremors in the extremities, and sensory and/or cerebellar ataxia [1,8,9]. Evidence of central nervous system demyelination can be found in up to 8% of these patients, configuring the central and peripheral demyelination syndrome [10,11]. Regarding treatment, these patients are characterized by poor response to intravenous immunoglobulin (IVIg) and other first-line immunotherapies such as corticosteroids and plasmapheresis. However, they usually respond adequately to B-cell depletion therapy, a feature linked to the IgG4 isotype of these antibodies [6,8].
We report the clinical characteristics of a patient diagnosed with chronic inflammatory demyelinating polyradiculoneuropathy anti-neurofascin-155, representing the first case reported in Peru, with a characteristic phenotype and striking findings such as signs of cerebellar affection and asymmetric postural tremor.
Case presentation
Patient’s history
A 38-year-old man with a history of bronchial asthma, chronic gastritis, and depression. Symptoms of the disease began eight months before admission, with paresthesias in hands and feet, initially episodic, which later became permanent. Subsequently, decreased sensibility in the lower limbs, predominantly distal, and gait instability appeared.
Seven months before admission, the patient noticed weakness in the lower limbs when walking long distances. Progressively, this made it difficult for him to climb stairs, associated with lumbar pain, initially localized, later radiating to both lower limbs. This pain partially subsided with anti-inflammatory drugs.
Four months before admission, weakness in the hands when performing fine movements was added. The weakness progressed to impairment in holding objects, also noting postural tremor in the left hand.
Three months before admission, the weakness in the lower limbs was pronounced, together with gait instability, which led to falls.
One month before admission, difficulty in articulating words, ingesting liquids, and diplopia were added. These new symptoms prompted the patient to seek care at our center (Figure 1).
Timeline.
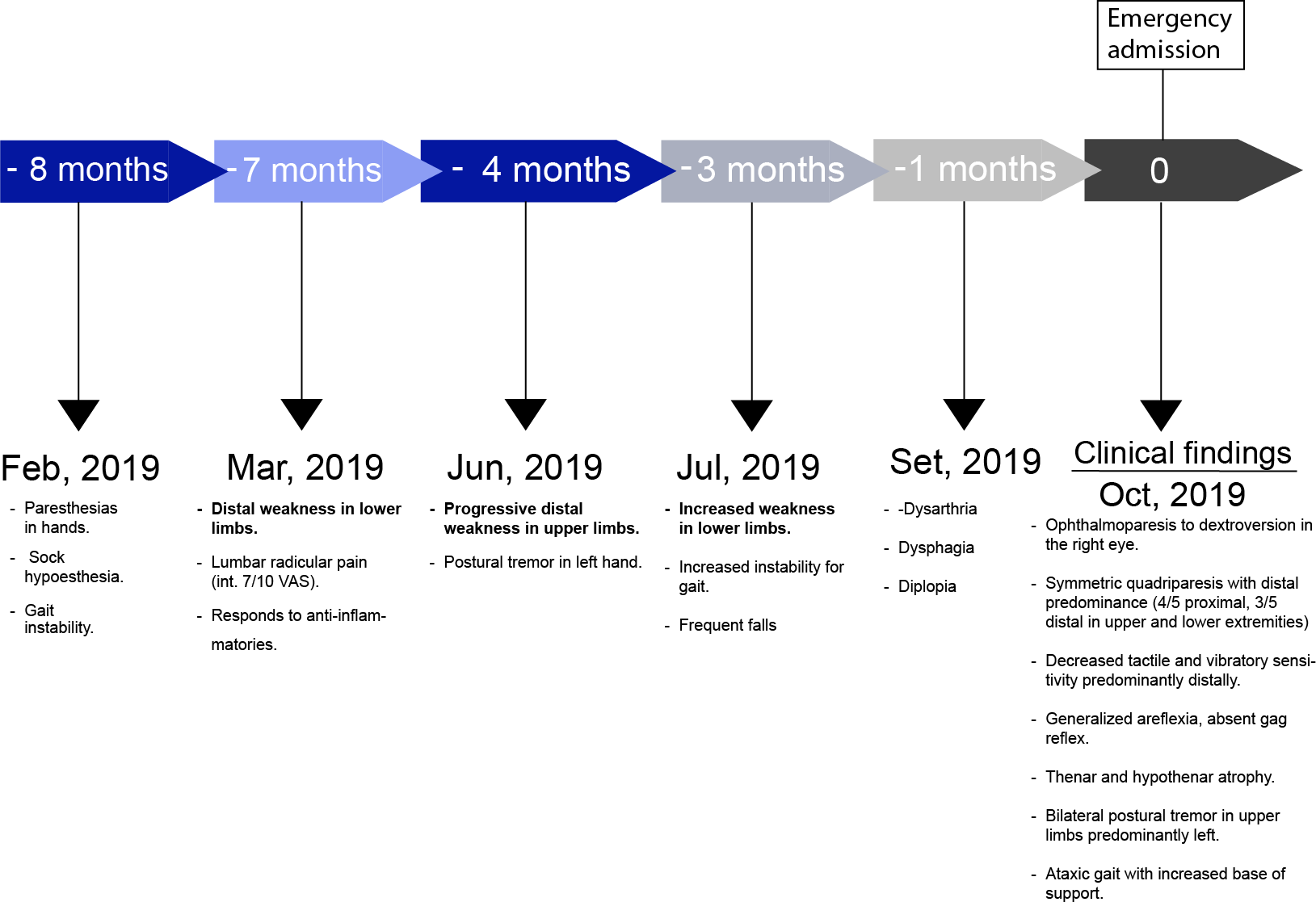
Source: Prepared by the authors of this study.
Clinical findings
Physical examination revealed an awake, lucid patient, with symmetrical facies, without respiratory impairment. Muscle strength evaluation showed distal predominant symmetrical quadriparesis, with a Medical Research Council (MRC) scale of 44 points: 4/5 for shoulder abduction, elbow flexion, hip flexion, and knee extension (right and left) and 3/5 for wrist and ankle dorsiflexion (right and left). The patient also presented decreased tactile and vibratory sensitivity, predominantly distal, and generalized areflexia; thenar and hypothenar hypotrophy; postural tremor of low frequency and high amplitude in upper limbs predominantly left; dysmetria and appendicular dyschronometry (index-nose and heel-knee test bilaterally altered), dysdiadochokinesia, absent gag reflex, ophthalmoparesis to dextroversion in the right eye and ataxic gait with an increased base of sustentation and positive Romberg’s sign.
Modified Rankin Scale: three.
Auxiliary diagnostic studies
Laboratory tests on admission showed: leukocytes: 4.42 per thousand units per liter; hemoglobin: 14.4 milligrams per deciliter; platelets: 282 per thousand units per liter; glucose: 110 milligrams per deciliter; urea: 32 milligrams per deciliter; creatinine: 0.82 milligrams per deciliter; enzyme-linked immunosorbent assay or HIV I/II adsorption enzyme immunoassay: non-reactive. Cerebrospinal fluid cytochemistry: protein: 533 milligrams per deciliter, cells: three per field.
Antinuclear antibodies, anti-neutrophil cytoplasm antibodies C and P, and antibody profile against extractable nuclear antigens: negative.
Vitamin B12 and folic acid: within normal values.
Electrophoretic proteinogram with immunofixation in blood: normal, anti-MAG antibody: negative.
Chest X-ray, thoracoabdominal-pelvic tomography, and brain MRI were normal.
Initial electromyography (taken in September 2019) was compatible with primarily demyelinating sensory-motor polyneuropathy(tables 1 and 2). Due to clinical suspicion, an anti-neurofascin-155 antibody test was requested and resulted positive.
Treatment and evolution
The patient was treated with methylprednisolone at a dose of one gram per day for five days, followed by one milligram per kilogram of prednisone for three months with progressive tapering. With this, the patient’s diplopia and dysphagia improved, with a slight improvement in limb strength. Control electromyography was performed six months after beginning corticosteroid therapy, which reported unexcitable potentials in all the evaluated nerves, both sensory and motor. Due to this, treatment with two grams of rituximab was started (one gram on day one and day 15). With this treatment, the patient presented remarkable improvement in distal muscle strength, tremor, coordination, and gait stability.
The control electromyography (carried out in October 2021), performed six months after the start of treatment with rituximab, showed recovery of all motor potentials but not of sensitive potentials, which remained undetectable(tables 1 and 2),
This report was approved by the National Institute of Neurological Sciences ethics committee, and the patient signed informed consent for publication of the case.
Discussion
We present the case of a 38-year-old male who presented with an eight-month history of paresthesia and progressive weakness of four limbs associated with diplopia and dysphagia. The prolongation of distal latencies, the marked decrease of conduction velocity in electromyography, and the presence of albuminocytological dissociation in cerebrospinal fluid supported the diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy, a variant of a distal symmetric acquired demyelinating neuropathy, due to its distal distribution.
In this context, we ruled out causes linked to this phenotype, such as anti-myelin-associated glycoprotein (anti-MAG) polyneuropathy, monoclonal gammopathies, and rheumatological diseases.
Once the previously mentioned causes were ruled out, an anti-neurofascin-155 (anti-NF-155) antibody study was requested since the patient presented atypical clinical features and predominantly distal involvement, such as the presence of tremor in the upper extremities and ataxia. The antibody test was positive, the first case with a serological diagnosis reported in Peru.
Chronic inflammatory demyelinating polyradiculoneuropathy is a clinically heterogeneous group of rare immune-mediated peripheral neuropathies that share neurophysiological manifestations of demyelination and albuminocytological dissociation; a hallmark of this entity, present in up to 80% of patients [1,12,13]. Within this clinically heterogeneous group, there is a typical variant, characterized by symmetric proximal-distal sensory-motor involvement of all four limbs in around 50% of cases, and atypical variants, including distal acquired symmetric demyelinating neuropathy, accounting for 7% of chronic inflammatory demyelinating polyradiculoneuropathy. This atypical variant is characterized by predominantly distal sensory-motor involvement (unlike typical chronic inflammatory demyelinating polyradiculoneuropathy), such as in our patient [14,15].
Several recently described antibodies directed against nodal and paranodal proteins, including anti-neurofascin-155, have allowed defining some atypical subtypes of chronic inflammatory demyelinating polyradiculoneuropathy, each with a specific clinical and treatment response profile [8].
Patients with anti-neurofascin-155 antibodies present with distinctive clinical features, such as predominantly distal sensory-motor affection (similar to the atypical variant of an acquired distal symmetric demyelinating neuropathy), early age of presentation, low-frequency and high-amplitude postural tremor, as well as sensory and cerebellar ataxia [8,9].
Our patient presented some features typically related to this phenotype, such as predominantly distal involvement, gait ataxia, and signs of cerebellar involvement, such as appendicular dysmetria and asymmetric postural tremor, similar to that reported by other authors [10]. Although an explanation for this asymmetry has not yet been established, we suggest that it could be linked to an immune response proportional to the variable expression of the antigen in various areas of the central and peripheral nervous system.
Up to 8% of patients with chronic inflammatory demyelinating polyradiculoneuropathy and anti-neurofascin-155 antibodies can present demyelinating lesions in the brain, which simulate multiple sclerosis lesions and, combined with chronic inflammatory demyelinating polyradiculoneuropathy, make up the central and peripheral demyelinating syndrome [9,12]. Our patient did not present encephalic lesions suggestive of this syndrome. Still, he presented clinical cerebellar impairment features such as dysmetria, dyschronometria, and appendicular dysdiadochokinesia, linked to the central nervous system expression of neurofascin-155, indicative of immune damage at this level that would escape the detection threshold of magnetic resonance imaging [5,8].
The acquired distal symmetrical demyelinating neuropathy variant may be the clinical expression of other chronic autoimmune neuropathies; up to 2/3 of patients with this pathology harbor an Ig M monoclonal gammopathy (acquired distal symmetrical demyelinating neuropathy-M). Of these, at least 50% express the antibodies against myelin-associated glycoprotein (MAG), which defines anti-myelin-associated glycoprotein neuropathy. The normal electrophoretic proteinogram and the absence of anti-myelin-associated glycoprotein antibodies allowed us to rule out these neuropathies, these being the main differential diagnoses [15].
Poor response to corticosteroids, plasmapheresis, and immunoglobulin and good response to B-cell depleting therapies such as rituximab is another characteristic of patients with anti-neurofascin155 antibodies due to the IgG4 nature of these antibodies [6,16].
Rituximab is an anti-CD20 monoclonal antibody used to treat these patients in different dosing regimens. The regimen of 375 milligrams per square meter weekly for four weeks, followed by a monthly dose for two additional months, is the most used. However, the two-gram schedule (one gram on day one and day 15) every six months has also been used with excellent results, although no studies have evaluated the efficacy of these dosing schedules [17].
Our patient responded only partially to initial corticosteroid therapy, improving diplopia and dysphagia, with no Medical Research Council and Modified Rankin variation, at six months. The electromyographic study at that time worsened from the initial study (in September 2019), as both sensitive and motor potentials became undetectable. In addition, the patient showed exacerbation of his gastritis due to corticosteroid consumption; thus, after the patient tested positive for anti-neurofascin-155 antibodies, we decided to escalate directly to rituximab treatment at doses of two grams (one gram on day one and day 15) every six months, with marked improvement in distal limb strength, tremor and gait stability.
After six months of rituximab treatment, they improved their Medical Research Council score to 60 points and their modified Rankin scale to one. This clinical improvement also correlated with a new electromyographic study (in October 2021, tables 1 and 2) where even recovery of previously unexcitable motor potentials was observed, without recovery of sensitive ones, similar to what was observed by other authors [18].
Conclusions
The case presented corresponds to an atypical subtype of chronic inflammatory demyelinating polyradiculoneuropathy associated with anti-neurofascin-155 antibodies. It was characterized by a phenotype with predominant distal sensory-motor involvement associated with asymmetrical low frequency and high amplitude tremor in the upper limbs, sensory ataxia, and signs of cerebellar affection, in addition to poor response to treatment with corticosteroids and excellent response to rituximab.
These features are linked to immunological damage in different regions of the central and peripheral nervous system with neurofascin-155 expression and IgG4 isotype of these antibodies.
