Case reports
← vista completaPublished on October 7, 2016 | http://doi.org/10.5867/medwave.2016.09.6568
Rare association of anophthalmia, complex congenital heart disease and pulmonary hypertension: case report
Rara asociación de anoftalmía, malformación cardíaca compleja e hipertensión pulmonar: reporte de caso
Abstract
Clinical congenital anophthalmia is described as the uni- or bilateral absence of the eyeball that might occur in isolation or as part of a syndrome. It has a very low prevalence and its etiology is heterogeneous. Complex congenital cardiac malformations are also rare. The association of congenital anophthalmia and congenital heart disease is rarer still, and the etiology of those associations is not well understood yet. We report the case of a patient who had the very rare association of bilateral anophthalmia, multiple cardiac malformations and severe pulmonary hypertension.
Introduction
Congenital clinical anophthalmia is described as the uni- or bilateral absence of the eyeball that can occur in isolation or as part of a syndrome. The prevalence of anophthalmia and microphthalmia is 3 and 14 per 100,000 births, respectively, while the combined prevalence might reach up to 30 cases per 100,000 births [1],[2],[3], and its etiology is heterogeneous. Although anophthalmia is clinically recognized, the biopsy can reveal residual ectoderm and, depending on the result, it can be classified as true anophthalmia, clinical anophthalmia or extreme microphthalmia. Some congenital heart diseases have a low prevalence and to see them together in a patient is very infrequent. The association of congenital malformations such as anophthalmia and complex heart disease has been rarely reported [1],[4],[5],[6],[7]. The etiology of such associations has not yet been fully elucidated. This is a case report of a patient who had the rare association of bilateral anophthalmia, multiple cardiac malformations and severe pulmonary arterial hypertension.
Case presentation
As policy of the research ethics committee of the hospital, the following report has informed consent signed by parents to submit and/or publish the data within the scientific context without showing the identity of the patient.
An 8-day-old female newborn delivered by normal spontaneous vaginal delivery after a full term pregnancy in a rural hospital was transferred and admitted in the Neonatal Intensive Care Unit. Her weight was 3.6 kg (p60), length 51 cm (p66), head circumference 35 cm (p66). The mother had a normal antenatal period and she reported no exposure to infections, alcohol, drugs or tobacco during pregnancy. Apgar scores were 5 and 7 at one and five minutes, respectively. The patient was the second born child of consanguineous parents (first cousins). The mother had a history of one miscarriage and a first female newborn that died minutes after the birth without a known cause.
On physical examination, the patient was hypoactive with a weak cry and in respiratory distress. The patient had dysmorphic facial features characterized by a narrow forehead, hypotelorism, small palpebral fissures, bilateral anophthalmia, depressed nasal bridge, anteverted nares and low seat ears. Other notable exam findings included right hand with preaxial polydactyly (bifid thumb), hirsutism and normal female external genitalia. Upon auscultation, a grade 4/6 systolic murmur was heard on the left superior sternal border. Patient had normal breath sounds with pulse oximetry of 85% despite supplemental oxygen by hood. After admission, patient had worsening respiratory distress that required assisted ventilation in synchronized intermittent mandatory ventilation mode.
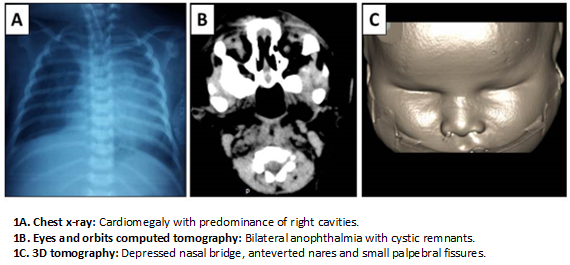
Chest radiograph was done and read as moderate cardiomegaly and pulmonary infiltrates probably related to congenital pneumonia (Figure 1A). It did not show any vertebral malformation. Abdominal ultrasound showed polysplenia and renal ultrasound was normal. Neonatal metabolic screening (hypothyroidism, congenital adrenal hyperplasia, phenylketonuria, galactosemia) and STORCH (syphilis, toxoplasmosis, rubella, cytomegalovirus and herpesvirus) were negative. Karyotype was 46xx.
Head computed tomography scan did not show any cerebral abnormality. Computed tomography of the eyes and orbits was read as absence of eyeballs and presence of cystic remnants, threadlike optic nerve, hypoplasia of extrinsic ocular muscles and cryptophthalmos (Figures 1B and 1C). Electroencephalogram showed disorganized sleep pattern without epileptiform activity.
 Full size
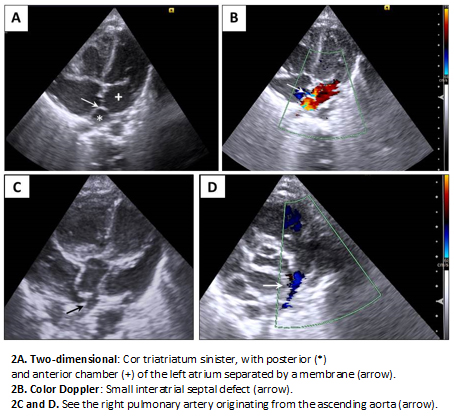
Full size Transthoracic echocardiography was read as situs solitus, cor triatriatum sinister (left atrium divided by a septum into an anterior and a posterior chamber) with moderate gradient across the membrane, atrial septal defect caudal to the membrane, hemitruncus arteriosus and signs of moderate pulmonary hypertension (Figure 2).
 Full size
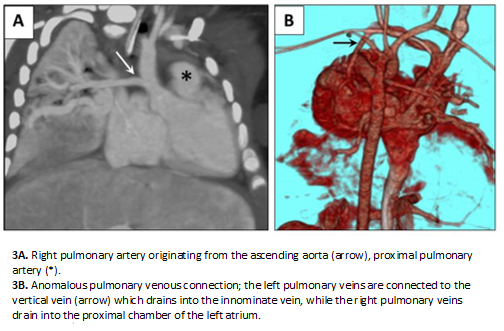
Full size Computed tomography angiography showed cor triatriatum sinister, a 4-milimeter atrial septal defect, partial anomalous pulmonary venous connection (cardiac and supracardiac), large patent ductus arteriosus, and left pulmonary artery originating from the ascending aorta (hemitruncus arteriosus). The origin of the left coronary artery was difficult to visualize (Figure 3).
 Full size
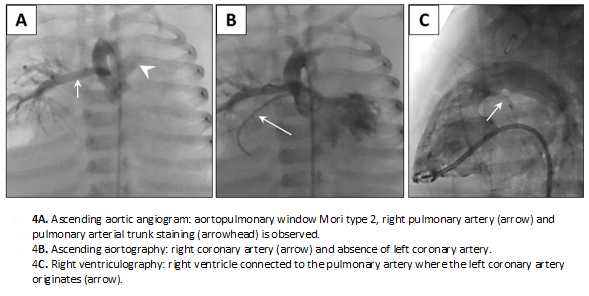
Full size Cardiac catheterization confirmed the large patent ductus arteriosus, cor triatriatum sinister and atrial septal defect. It also showed a Mori type II aortopulmonary window, and a partial anomalous pulmonary venous connection: left pulmonary veins were seen draining into the innominate vein by way of the vertical vein, while the right pulmonary veins were seen draining into the proximal chamber of the left atrium. Also noted were a juxta-ductal aortic coarctation, anomalous origin of the left coronary artery from the pulmonary trunk (ALCAPA or Bland-White-Garland Syndrome) (Figure 4), hypoplasia of the pulmonary arteries with Nakata index 110 mm2/m2 (normal value: 330 +/- 30 mm2/m2) [8] and severe pulmonary hypertension with negative vasoreactivity testing.
 Full size
Full size At six months of age, tracheostomy and G-tube placement were done. Patient was transferred to the Pediatric Intensive Care Unit. During hospitalization, patient received various antibiotic treatments for recurrent pneumonias, most likely related to prolonged ventilation.
The patient did not have surgery because of the complexity of the congenital heart disease. Although the hospital arranged all the necessary paperwork to transfer the patient to a cardiology center, it was unsuccessful. Patient died after a cardiac arrest at seven months of age. Parents did not give authorization for an autopsy.
Discussion
Anophthalmia can appear as an isolated physical feature but it may also be part of a variety of syndromes such as: Lenz, Fraser, Matthew-Wood syndrome (Spear syndrome or PDAC: pulmonary hypoplasia/agenesis, diaphragmatic hernia/eventration, anophthalmia/microphthalmia and cardiac defect), oculofaciocardiodental syndrome, anophthalmia-esophageal-genital syndrome and CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital abnormality, and ear abnormality) syndrome. These syndromes can be caused by genetic disorders that can be inherited in a dominant fashion (SOX2 mutations) or in a recessive fashion (CHX10 mutations). Anophthalmia may also be associated with infections during pregnancy, such as with cytomegalovirus infection [2],[3],[9],[10]. The association between anophthalmia, congenital heart disease and pulmonary hypertension is very rare and its etiology is not yet fully understood.
As in other countries of the region, the lack of technological resources makes diagnostic work-up in cases like these difficult and genetic counseling challenging. In this particular case, the history of consanguinity and previous miscarriage were highly suggestive of an inherited disease, making it likely for this couple to have a new descendant with similar features in the future.
Even with the diagnosis of pulmonary hypertension, vasodilators were not prescribed to this patient. There was a high risk of causing coronary steal if the pulmonary vascular resistances were diminished. Risk of provoking acute heart failure outweighed the benefits of administering the medication.
Conclusions
The spectrum of anophthalmia and congenital heart defects is wide and diverse. There are some established syndromes in which these phenotypes are associated. However, the congenital heart defects described in this patient have been previously reported only as separate entities and not as a part of a syndrome such as the one being described in this case report. The present case report adheres to the CARE guidelines for clinical case reports [11].
Notes
From the editor
The authors originally submitted this article in Spanish and English. The Journal has not copyedited the English version.
Ethical aspects
The father of the patient signed the informed consent requested by Medwave. A copy was sent to the Journal editorial board.
Conflicts of interest
The authors completed the ICMJE conflict of interest declaration form, translated to Spanish by Medwave, and declare not having received funding for the preparation of this report, not having any financial relationships with organizations that could have interests in the published article in the last three years, and not having other relations or activities that might influence the article´s content. Forms can be requested to the responsible author or the editorial direction of the Journal.
Funding
The authors declare that there was no funding coming from external sources.
