Case reports
← vista completaPublished on April 29, 2020 | http://doi.org/10.5867/medwave.2020.03.7880
Progressive hemifacial atrophy or Parry-Romberg syndrome: A pediatric case report
Atrofia hemifacial progresiva o síndrome de Parry-Romberg: reporte de caso pediátrico
Abstract
Progressive hemifacial atrophy—or Parry-Romberg syndrome—is a rare disease, classified as one of the forms of localized morphea or scleroderma. Its cause is unknown. It is characterized by atrophy of the skin, fat, muscles and underlying osteocartilaginous structures that usually affects the face and neck unilaterally, and is associated with neurological symptoms (secondary epilepsy) and involvement of other organs and systems. Its course is slow and progressive and begins in the first two decades of life. Predilection for female sex has been observed. We report the case of a 10-year-old girl diagnosed at the Hipólito Unánue Hospital in Tacna, Peru. Knowledge of this condition is important in the differential diagnosis of localized morpheas or scleroderma.
Key ideas
|
Introduction
Progressive hemifacial atrophy, also known as Parry-Romberg syndrome, was first described by Caleb Hillier Parry and Moritiz Heinrich Romberg in 1825 and 1846, and is characterized by progressive atrophy of skin and underlying tissues, affecting the face and neck usually unilaterally[1],[2],[3],[18]. Its incidence is about three cases per 100 000 people per year[4]. Although this rare disease normally only affects the face and neck unilaterally, it occasionally manifests bilaterally and even affects the trunk, arms, and legs. Its course is progressive, slow and arises mainly between the ages of two and 20 years[5],[6],[7].
The pathogenesis of this disease is unknown[8],[11]; nevertheless, a variety of theories have been proposed, including immunological, traumatic[1], infectious, endocrinological[12], neurological, and genetic causes[2]. Epilepsy is the most frequent anomaly associated with Perry-Romberg syndrome.
We report a 10-year-old girl diagnosed with progressive hemifacial atrophy at the Hipolito Unanue Hospital in Tacna, Peru, in 2015. As far we know, this is the third case reported in Peru and the first case in Tacna[13].
Case report
This is the case of a 10-year-old female patient from Tacna (a city in southern Peru bordering Bolivia and Chile). She is the product of a second gestation and was born at term by cesarean delivery. Her parents are not blood related and her immediate family members are presumed healthy. In the perinatal period, no relevant disease was detected. The physical and motor development was adequate.
Our patient came to the emergency department of Hipólito Unánue Hospital presenting loss of consciousness and tonic-clonic seizures that evolved to focal epileptic state. For this reason, the girl was admitted for further studies and treatment.
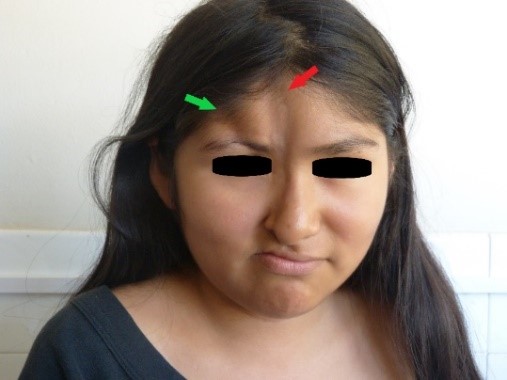
At the physical examination, we observed the presence of two atrophic lesions in the face: the first and longest lesion, located at the facial medial line, was eight centimeters in length, two centimeters in width, and between one and two millimeters in depth, spreading from the trichion to the right wing of the nose; mild hyperpigmentation was noticed at its apex (Figures 1 and 2, indicated by red arrow). The second atrophic lesion, located laterally from the first lesion, spread from the right frontal hairline to the right supraciliary arch, where a zone of madarosis was observed, and was four centimeters in length, one centimeter in width and seven millimeters in depth (Figures 1 and 2, lesion indicated by green arrow).
 Full size
Full size 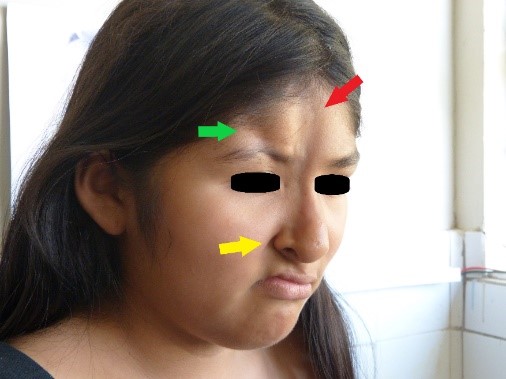 Full size
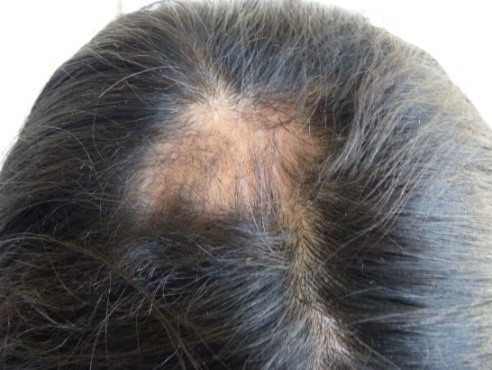
Full size In the right parietal region, an alopecia zone was found (Figure 3). At the same time, we observed anisocoria and mydriasis of right eye pupil, but it was still photo reactive. Also noted were moderate right palpebral ptosis, right corneal ulcer, dislocation of the right lens and ametropy. No abnormalities were found in the retina. Atrophy of right wing of the nose was noted, along with a deviated septum, elevated right oral commissure, right nasogenian sulcus accentuation (Figure 2, yellow arrow), and abnormal position of teeth (Figure 4). The rest of the examination was normal.
 Full size
Full size 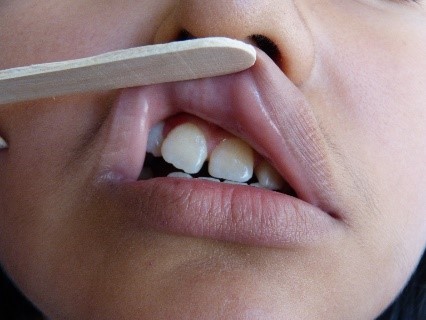 Full size
Full size At the age of three years old, our patient suffered brain trauma with loss of consciousness and seizures. At that time, these symptoms were assumed to be caused by the trauma. One year later, her parents noticed the presence and progression of the abnormality of the face (described in the paragraphs above); however, they did not look for medical assistance because they assumed it was a consequence of the previous trauma.
The laboratory exams that we performed for the patient showed mild leukocytosis with neutrophilia (18.3 x 103cell per mm3, 79% neutrophils) and elevated glycemia (149.8mg/dl). These findings were explained by seizures. We obtained negative (normal) results for testing of anti-nuclear antibodies, anti-neutrophil cytoplasmic antibodies, hepatitis B surface antigen, and IgM against hepatitis A virus.
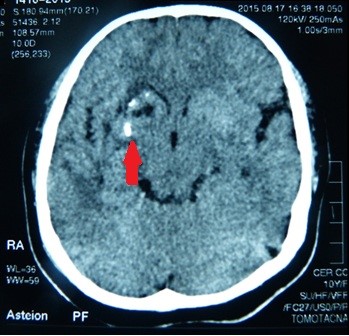
The brain computed tomography (CT) scan showed calcifications and surrounding edema in the right basal ganglia: in the putamen and in the head of caudate nucleus (Figure 5). In the 3D CT reconstruction of the skull, we found asymmetry of the facial bones: atrophy of right maxillary bone and of the frontal bone, abnormal placement of teeth, nasal septum deviation and overgrowth of the lower left turbinate (Figure 6).
 Full size
Full size 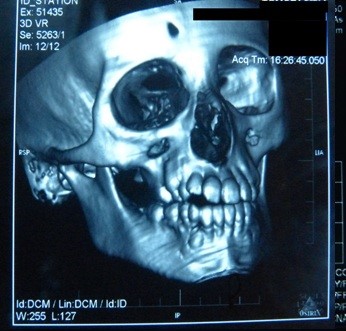 Full size
Full size Once we obtained the data from the anamnesis, physical examination, and diagnostic tests, the main author of this article carried out an exhaustive search in databases of rare diseases (such as Mendelian Inheritance Online in Man - OMIM, available at www.omim.org), concluding that the girl's condition was compatible with Parry-Romberg syndrome or progressive hemifacial atrophy.
Having achieved adequate control of seizures, the patient was discharged from the hospital; however, she is under medical observation by a neurologist, pediatrician, ophthalmologist, and psychologist. This multidisciplinary team has the aim to improve the quality of life of our patient.
Discussion
Parry-Romberg syndrome is a rare disease characterized by progressive atrophy of the skin, muscles, and bones that compromises the face, usually unilaterally[4],[18], and in some cases is accompanied by abnormality of dentition, tongue, and palate[1],[7]. Although Parry-Romberg syndrome is usually unilateral, it is possible to find patients with bilateral damage, even finding those with compromised trunk, arms, and legs[4].
The incidence of this disease is three cases per 100 000 patients per year[4] and is more common in women with proportion of 3:1. It typically presents in the first twenty years of life[1].
This disease is usually classified as lineal localized esclerodermia or morphea. In world literature, we found references to Parry-Romberg syndrome as morphea in coup de sabre (“saber strike”, because of the similitude of skin atrophy to the wound caused by saber). However, from our understanding, these conditions are two different entities, as the patients suffering Parry-Romberg syndrome do not show histological features of dermic sclerosis, meanwhile patients with morphea in coup de sabre do show it[9],[10].
The etiology of Parry-Romberg syndrome is still unknown; however, the most accepted hypothesis is the autoimmune etiology[1],[7]. The Th1 and Th17 inflammatory pathways are of great importance in the pathophysiology of this disease, at least in the early stages of it. The Th2 inflammatory pathway is important in the late stage, the fibrotic stage. This inflammatory response of the tissues could be initiated by an incidental traumatic event, surgery, or even obstetric trauma[7],[23]. Neurological theory suggests that this disease is the result of disorganized migration of neural crest cells that compromises one or more of the branches of the trigeminal nerve[24].
Some reports claim that Parry-Romberg syndrome is a genetic autosomal dominant disease. Moreover, it has been said that progressive hemifacial atrophy is linked to Lyme disease[14], Borrelia burgdorferi, and to another infections like syphilis, rubella, tuberculosis[7] and the hepatitis B virus[15].
In this case, the patient developed the illness after a fall, acquiring head trauma. There were no prior records of illness in the family. Her laboratory tests for presence of anti-nuclear antibodies and viral hepatitis were negative. We did not evaluate our patient for Lyme disease because Peru is not an endemic area for this disease, and she denied having traveled to endemic regions.
The Parry-Romberg syndrome involves dermatomes of one or more branches of the trigeminal nerve and usually compromises the left half of the face[2],[18]. It could present some grade of increase of pigmentation and brightness of the skin. Alopecia can be seen in any area of scalp but usually in the frontoparietal region[1],[7].
M. Wong, in his case series, demonstrates a disease predilection for the left half of the face[2]. Carlos Galarza in his two cases reported from Cuzco, Peru, also demonstrated skin atrophy on the left side[20]. Meanwhile, our patient presented skin atrophy located in the right half of face, which extended from frontal to mandibular regions, accompanied by mild hyperpigmentation, septum deviation, abnormal distribution of teeth, and parietal location of alopecia.
Ocular manifestations of Parry-Romberg syndrome may be present before, during, or after the appearance of skin atrophy, and may include: enophthalmos leading to diplopy (caused by fat atrophy in retro bulbar space), dry eye, or uveitis. There have also been reports of eyelid pigmentation, photophobia, strabismus, iris atrophy, panuveitis, hypotonic ciliary body, retinal vasculitis, retinal edema and detachment, etc[4],[16].
Neurological evaluation may show oculomotor nerve palsy manifesting with pupillary abnormalities, anisocoria, mydriasis or myosis on the affected side and involvement of optic nerve that can lead to neuroretinitis and papillitis[16]. Our patient, since her first visit to the emergency department, suffered severe anisocoria and mydriasis of the right eye, an abnormality of pupillary reaction, and as the disease progressed, other manifestations appeared: dislocation of right lens, corneal ulcer, severe enophthalmos, and ametropy.
Dalla Costa, in her worldwide review of 205 patients diagnosed with Parry-Romberg syndrome, reported that 50% of patients had symptoms of a compromised central nervous system and 15% had epileptic seizures, headaches, facial pain, cranial nerves involvement and hemiplegia[17].
The most frequent neurological manifestation of Parry-Romberg syndrome is epilepsy (60.5% frequency). Focal seizures ipsilaterally to brain calcifications are common (seen in 50% of total cases). With a frequency of 33%, secondary epilepsy is very difficult to treat. Headache (44%) and trigeminal neuralgia (8.5%) may also be present[4].
Intraparenchymal brain calcifications on the same side as facial abnormalities are the most frequent findings in computer tomography and magnetic resonance imaging. White matter hyperintensities, focal infraction of the corpus callosum, cerebral hemiatrophy and leptomeningeal enhancement[3],[7], intracranial aneurysms and vascular malformations[3] and brain microhemorrhages[4] have been described. There is no correlation between neuroimaging and facial features of this disease[19].
Although neurological and ocular characteristics are the most frequent manifestations of Parry-Romberg syndrome, it is possible to find cardiac abnormalities (hypertrophic cardiomyopathy), endocrine abnormalities (hyperthyroidism, hypothyroidism), autoimmune diseases (primary biliary cirrhosis, rheumatoid arthritis, multiple sclerosis), and congenital diseases, such as Poland syndrome, microphthalmy, and kidney malformations[4].
Secondary epilepsy and headaches were the predominant neurologic features in our case. Epileptic seizures had been well controlled. Tomography and resonance results were described as calcifications in basal ganglia: putamen and nucleus caudate. We did not find any other abnormality beyond those described above.
The goal of the treatment is to stop the active phase of disease. This can be achieved with the prescription of methotrexate at 0.3 – 1mg/kg/week for one to two years, and it can be associated with prednisone during the first three months considering the delayed effect of methotrexate on inflammation and fibrosis[21].
Parry-Romberg syndrome has a great biopsychosocial impact on the patient's life due to functional and aesthetic limitations. Some surgical approaches for restoring facial symmetry have been proposed. Mild and moderate cases can be treated with silicone fillers, collagen, porous polyethylene implants, and autologous fat grafting. The grafting of cartilage, bones, fat, and skin are treatment options for more severe cases[21],[22]. The aim of psychological treatment[22] is the social reintegration of patients.
The prognosis of the disease depends on the age of the patient, being more severe when atrophic changes begin in adulthood (aged 20 and older). The severity of atrophy, brain damage, and poor response to treatment are higher in patients older than 20[18]. These determinants give us the global approach to the sequelae and the treatment that must be considered to obtain better therapeutic results[20].
Conclusions
Progressive facial hemi atrophy, or Parry-Romberg syndrome, is a rare, progressive disease of unknown cause that mostly affects women. There are few reported cases of this syndrome in Peru, and this is the first case in the Tacna region.
It is characterized by the progression of atrophy, usually unilateral, being more frequent on the left side of the face and involving osteocartilaginous structures. In our case, the skin abnormality was found on the right side; this is an infrequent finding.
Knowledge of this disease becomes relevant in the differential diagnosis of atrophic lesions in one side of the face. Among the diseases with which the differential diagnosis must be made, the following stand out: coup de sabre disease, deep lupus, steroid atrophy, and other types of morphea.
Notes
Author contributions
EOL: main author, diagnosis, and follow-up of the patient, writing and critical review of the article, final approval of the article and assumption of responsibility for the manuscript. SDA: author, writing and important contributions to the article and assumption of responsibility for the manuscript. PDA: co-author, important contributions to the article
Conflict of interests
None of the authors declare having conflicts of interest with the subject of this article.
Funding source
The financing in the elaboration of the article is own.
Ethical aspects
The authors have obtained the informed consent of the patient's parents (being that she is a minor) for the publication of this article and the accompanying images.
Data
We declare availability for the delivery of data on request.
From the editors
The original version of this manuscript was submitted in Spanish. This English version was submitted by the authors and has been lightly copyediting by the Journal.
