Research papers
← vista completaPublished on October 21, 2021 | http://doi.org/10.5867/medwave.2021.09.8474
Effectiveness of biosimilar drugs in the treatment of renal anemia: A case series
Efectividad del uso de biosimilares en el tratamiento de la anemia en pacientes con enfermedad renal crónica: serie de casos
Abstract
Introduction Patients with chronic kidney disease usually have anemia secondary to an erythropoietin deficit. The emergence of biosimilar drugs of erythropoiesis-stimulating agents ensures broader access to these treatments.
Objective This study analyzes the effectiveness of an epoetin α biosimilar drug in chronic kidney disease patients with anemia.
Methods This observational retrospective study enrolled 111 consecutive outpatients with chronic kidney disease and anemia and criteria for using erythropoiesis-stimulating agents. We collected baseline epidemiological and comorbidity data, as well as hematological and renal function infor-mation. We analyzed the effectiveness of the biosimilar agent in naïve patients and those who already had other erythropoiesis-stimulating agents.
Results The 111 included patients had a mean age of 83 ± 8 years, and 54% were males. We found that patients who previously received erythropoiesis-stimulating agents, maintained hemoglobin values at two months of treatment with the biosimilar, while the naïve group significantly raised their hemoglobin values (P < 0.001). Renal function remained stable within the whole sample. The cost of using erythropoiesis-stimulating agents was reduced by a mean of 82 ± 17% with the biosimilar drug.
Conclusion Using a biosimilar of epoetin α is effective in patients with chronic kidney disease and anemia and significantly reduces costs.
|
Main messages
|
Introduction
Anemia is commonly seen in patients with chronic kidney disease. This complication is usually due to multiple etiologies, and its magnitude increases as the glomerular filtration rate deteriorate [1]. There are multiple causes of anemia within patients with chronic kidney disease that include: iron deficiency, vitamin B12 deficiency, folic acid deficiency, uremic toxins, alterations in mineral metabolism, inflammation or myelotoxic drugs, and – above all – a decreased capacity for renal production of erythropoietin [2]. To alleviate this situation, biological drugs such as erythropoiesis-stimulating agents are currently available. Since their emergence, erythropoiesis-stimulating agents have improved patients' quality of life, reduced the number of transfusions, and even reduced mortality and hospitalizations in patients with chronic kidney disease and anemia [3],[4]. However, these agents are not exempt from adverse effects, such as an increased cardiovascular risk, hypertension, and thrombosis [5].
Biologic drugs have a high cost derived from their intense development and evaluation, generating inequity among patients. In 2008, the first epoetin α biosimilar drug was marketed in Spain. Biosimilar drugs are products similar to the original molecule but with a specific and independent development, which is never identical. Therefore, these drugs must demonstrate their non-inferiority to the reference drug [6]. The European Medicines Agency has carried out the regulation of the approval of biosimilar drugs since 2005. It includes the performance of phase one and three clinical trials before their launch [7]. To date, there are more than 60 biosimilar drugs marketed in Europe.
Regarding epoetin α biosimilars, some clinical trials have demonstrated their safety profile in phase one studies and their effectiveness in phase three studies [8],[9]. To date, post-authorization studies aimed at re-evaluating the long-term effectiveness and safety of these drugs are ongoing. However, real-life observational studies already published have some limitations. In general terms, they have heterogeneous inclusion criteria of patients with and without chronic kidney disease and analyze the effectiveness of biosimilars, without evidence on specific agents among specific populations. Concerning epoetin α, the available studies are scarce, both for biosimilar initiation and switch from other erythropoiesis-stimulating agents.
The present study aims to evaluate the use of a biosimilar drug of epoetin α in a clinical practice context among patients with chronic kidney disease. For this purpose, its effectiveness and efficiency were analyzed among patients not receiving erythropoiesis-stimulating agents and patients already treated with them.
Methods
Study design
We conducted a retrospective observational study, including outpatients in which an epoetin α biosimilar drug was initiated at our center (Hospital Universitario de La Princesa) in 2018.
Participants
We included patients over 18 years of age, with stage three or higher chronic kidney disease, that had criteria for initiation of erythropoiesis-stimulating agents or who were already on treatment with them and maintained these criteria. To define criteria, we relied on the Kidney Disease Improving Global Outcomes (KDIGO) guidelines for anemia and the National Institute for Health and Care Excellence (NICE) published in 2012 and 2015, respectively [10],[11]. We initiated erythropoiesis-stimulating agents in patients with hemoglobin values below 10.5 grams per deciliter, aiming to maintain a range between 10.5 and 11.5 grams per deciliter and avoid increases greater than 2 grams per deciliter in one month. We excluded patients with iron, folic acid, and vitamin B12 deficiency, patients admitted to a hospital during the study or within two months before starting the drug, or who had any biosimilar drug contraindication. Patients without complete clinical data were also excluded.
Variables
We used previous mentioned guidelines to define absolute iron deficiency as a transferrin saturation index lower than 20% and ferritin lower than 100 micrograms per deciliter. On the other hand, functional iron deficiency was defined as a transferrin saturation index lower than 30% and a ferritin value lower than 500 micrograms per deciliter.
Demographics, renal function (creatinine and glomerular filtration rate estimated by the Chronic Kidney Disease Epidemiology Collaboration, CKD-EPI), hemoglobin, transferrin saturation index, ferritin, and previous iron use were collected at baseline [12]. We classified patients into chronic kidney disease stages based on the estimated glomerular filtration rate [13].
Among patients on treatment with erythropoiesis-stimulating agents, we collected the agent they were using and their dose. We used a conversion factor to change from erythropoiesis-stimulating agents to the biosimilar drug. This formula consisted of maintaining the dose in the case of epoetin β, multiplying by 200 the weekly dose in micrograms of darbepoetin α, and multiplying by 800 the dose of methoxy-polyethylene glycol epoetin β (also in micrograms) [14].
The collected variables were compared in those who received erythropoiesis-stimulating agents versus those who did not.
Sources
The electronic medical record was used as a source of information.
Bias
Potential design (observational), confounding, and selection biases were mitigated by including a homogeneous and complete population.
Sample size
It was determined using the clinical registry, including all patients needing erythropoiesis-stimulating agents and who had received a biosimilar.
Qualitative (outcome) variables
Patients were evaluated two months after starting treatment with the biosimilar to analyze hemoglobin and renal function changes considering the clinical guidelines, records, and the clinical practice protocols used in our environment.
Statistical methods
Variables are expressed as mean ± standard deviation if they follow a normal distribution (according to the Kolmogorov-Smirnov test) or as median (interquartile range) otherwise. For statistical inference, we used the Chi-square test or Fisher's test and Student's t-test or Mann-Whitney test according to the normality results to compare the baseline characteristics between the groups with and without erythropoiesis-stimulating agents at two months of follow-up. We performed a complete economic evaluation, considering clinical outcomes and costs, and using a cost-effectiveness-incremental analysis [15].
All statistical analyses were performed with SPSS software (SPSS Inc., Chicago, IL) version 21.0. A P value < 0.05 was considered statistically significant.
Results
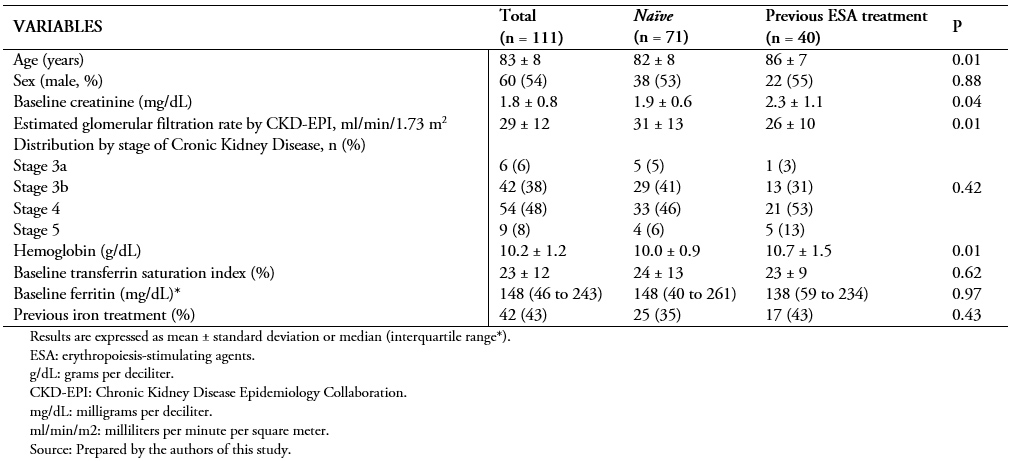
We included 111 patients from nephrology outpatient clinics, of whom 60 (54%) were male with a mean age of 83 ± 8 years. The distribution by stage of chronic kidney disease showed that most patients had stage 3b (42.38%) or 4 (54.48%). Mean creatinine was 1.8 ± 0.8 milligrams per deciliter, and glomerular filtration rate estimated by CKD-EPI was 29 ± 12 milliliters per minute per 1.73 square meters. As for hematological parameters, the mean hemoglobin was 10.2 ± 1.2 grams per deciliter. The rest of the baseline characteristics are shown in Table 1.
 Full size
Full size Seventy-one patients (64%) were not receiving any treatment with erythropoiesis-stimulating agents at baseline. From 40 (36%) patients receiving some type of erythropoiesis-stimulating agent, most were administered with methoxy-polyethylene glycol epoetin β (72.5%), followed by darbepoetin α (25%) and only one patient with epoetin β (2.5%). Table 2 specifies the mean baseline doses of each drug.
 Full size
Full size We performed a comparison between patients who received erythropoiesis-stimulating agents and those who did not. We found significant differences in age, baseline creatinine and glomerular filtration rate, and hemoglobin values between groups.
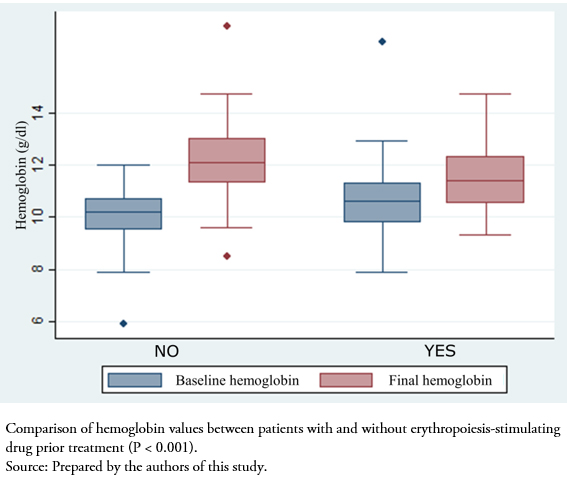
After initiating treatment with the biosimilar drug, we analyzed the variation in hematological and renal function parameters at 2 months. Naïve patients to erythropoiesis-stimulating agents had significantly increased hemoglobin levels after initiation of the biosimilar (difference between baseline and two months: 2.2 ± 1.4 grams per deciliter, P < 0.001). As expected, in patients who were switched from other erythropoiesis-stimulating agents, no differences were found after initiation of the biosimilar (0.6 ± 1.8 grams per deciliter, P > 0.05). There was no difference in the variation of glomerular filtration rate estimated by CKD-EPI between naïve patients and those receiving erythropoiesis-stimulating agents (+1.7 ± 6.2 versus -0.1 ± 5.7, P = 0.23) (Figure 1).
 Full size
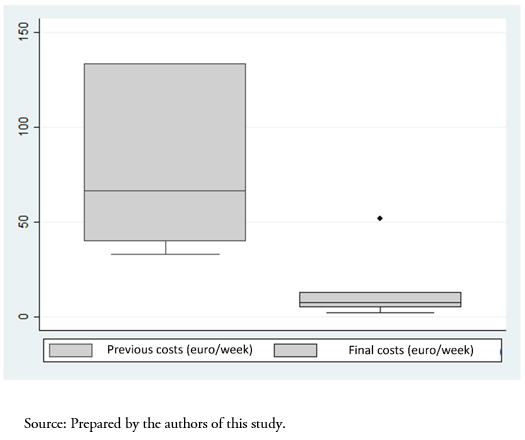
Full size Switching to the biosimilar resulted in a cost reduction of an average of 83 ± 11% compared to the initial price (previous cost 81 ± 41 versus 10 ± 7 euros per week, P < 0.001) (Figure 2). Table 3 shows the complete economic evaluation in both study groups, demonstrating a saving of 131 euros per unit of hemoglobin and patient using the biosimilar.
 Full size
Full size 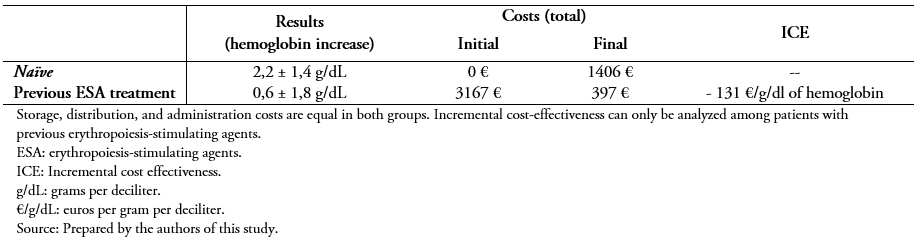 Full size
Full size No adverse effects were recorded with the use of the epoetin α biosimilar.
Discussion
For the first time in a clinical practice context, results confirm that patients with anemia and chronic kidney disease without treatment with erythropoiesis-stimulating agents benefit in terms of hemoglobin when receiving a biosimilar, with an excellent safety profile. In addition, those already receiving treatment with erythropoiesis-stimulating agents maintain their hemoglobin levels after switching to the biosimilar but achieve significant efficiency. This significant cost saving has been shown to allow greater access to biological treatments, which has a significant clinical impact. Specifically, our real-life data reflect a reduction of more than 80% of costs in patients switched to a biosimilar drug. This improved efficiency had already been demonstrated in a published paper on a cost model, theoretically using a patient with cancer-induced anemia in different regions of the world, although not as strong as our data demonstrate [16].
We found a mean increase of 2.2 ± 1.4 grams per deciliter of hemoglobin values at a 2-month follow-up among patients who were not receiving erythropoiesis-stimulating agents. The use of epoetin α biosimilars in chronic kidney disease has been reported in different studies with similar results. One of the few studies revealing quantitative data on the effectiveness of biosimilars was derived from the Italian Treviso registry published by Ingrasciotta et al. [17]. This study incorporated all patients who had received any epoetin biosimilar, including those with chronic kidney disease and cancer. At three months of treatment, the increase in hemoglobin values was significant in patients with chronic kidney disease (1.5 ± 1.1 grams per deciliter), with no differences compared with patients receiving the reference drug (1.6 ± 1.1 grams per deciliter). It should be noted that the response to erythropoiesis-stimulating agents has multiple factors involved, including inflammation, uremia, neoplasms, nutrition, or pharmacological interactions. Even though patients in our study responded homogeneously to biosimilars, resistance to erythropoiesis-stimulating agents should be further evaluated and treated to avoid risks related to overdosing [18].
In a subgroup of patients already receiving erythropoiesis-stimulating agents, we analyzed the variation of hemoglobin values after switching to the biosimilar. After two months of follow-up, we found no differences in hemoglobin levels before and after the switch. A propensity score-matched study published by Belleudi et al. analyzed the effectiveness and safety of switching from an erythropoiesis-stimulating agent to a biosimilar. Based on a biosimilar drug study network (ItaBioNet), authors obtained information of 14 400 incident patients on the use of erythropoiesis-stimulating agents for anemia, of whom 1866 switched to a biosimilar [19]. In terms of effectiveness, the results concluded a non-inferiority of the biosimilar drug with similar adverse effects compared with patients receiving erythropoiesis-stimulating agents [5].
Although age is considered a predictor of lack of response to erythropoiesis-stimulating agents, the mean age of the patients in our study was 83 ± 8 years, exceeding that of previously published studies and demonstrating the effectiveness of the drug even among aged populations [20]. Furthermore, we found differences in the mean age of both groups (naïve and those already receiving erythropoiesis-stimulating agents), without this implying a change in the expected response to the use of the biosimilar.
Regarding safety, we did not find any relevant adverse effect after two months of follow-up. The post-authorization study performed with erythropoiesis-stimulating agents (EPO-PASS) revealed the expected adverse reactions. Furthermore, the authors found no patient with antibodies against epoetin or pure red cell aplasia [21].
Our study is not without limitations. First, the retrospective and observational design come with limitations, such as data loss during follow-up or the use of medical records as a source. However, we followed the recommendations of clinical guidelines, and therefore, the patients presented serial hemoglobin determinations. In addition, those patients in whom we did not have clinical information regarding effectiveness were excluded from the study to have a more homogeneous population. Second, the sample size and lack of follow-up were also limitations. However, the results are conclusive for analyzing effectiveness and efficiency, considering that this study is the largest published to date. The decision to set a two-month evaluation point is based on clinical guidelines and the drug's technical data sheet, which coincides with our usual clinical practice [10],[11]. This evaluation allowed us to determine the results produced by the biosimilar in stable patients in a short-term analysis. In any case, we must be cautious with the extrapolation of data from observational studies due to their inherent limitations. Third, the safety results only refer to the first two months after drug initiation, so longer-term studies are needed to verify this situation in clinical practice. And finally, we found expected differences between the two study groups in their baseline characteristics (age, hemoglobin, and renal function). However, we do not believe they interfere with the study results. These differences are reasonable given that some patients underwent previous treatment with erythropoiesis-stimulating agents, and therefore, had more renal dysfunction and better hemoglobin levels.
Conclusion
The use of an epoetin α biosimilar drug effectively increases hemoglobin levels in patients without previous treatment with erythropoiesis-stimulating agents. Moreover, it represents a significant cost reduction compared to the reference drug.
The biosimilar drug field is still underdeveloped, so clinical research progress is needed for a universal use of this drugs. Undoubtedly, our results provide new evidence for biosimilars, but this information warrants subsequent longer-term studies addressing the importance of efficiency.
Notes
Contributor roles
PMR: study design, data acquisition, review, and approval of the final version. YGG: data acquisition, review, and approval of the final version. VAC: study design, article writing. BQ: study design, data acquisition, analysis and interpretation, article writing.
Competing interests
BQ and VA have participated in paid papers by Sandoz with the content of the present research.
Funding
The authors declare that they have not received funding for the present work.
Ethics
The study was approved by the Clinical Research Ethics Committee of Cantabria, obtaining the adequacy of the Hospital Universitario de la Princesa on October 9, 2017.
Data sharing statement
Data delivery could be available upon request.
Language of submission
Spanish.
