Publicado el 1 de septiembre de 2009 | http://doi.org/10.5867/medwave.2009.09.4153
Actualización en enfermedad de Kawasaki
Update on Kawasaki disease
Resumen
Este texto completo es una transcripción editada de la conferencia que se dictó en el XLVIII Congreso Chileno de Pediatría realizado en Viña del Mar entre el 26 y el 29 de Noviembre de 2008. El congreso fue organizado por la Sociedad Chilena de Pediatría bajo la presidencia de la Dra. Lidya Tellerías C.
Historia de la enfermedad de Kawasaki
La enfermedad de Kawasaki (EK) tiene una presentación clínica compleja y no se ha logrado demostrar su etiología, por lo tanto no se sabe cómo prevenirla; sin embargo existe un tratamiento efectivo.
Kawasaki describió por primera vez la enfermedad que lleva su nombre en 1967, como un síndrome febril óculo-oro-cutáneo, acrodescamativo, con descamación de la piel alrededor de uñas, palmas y plantas, con o sin linfadenitis cervical aguda no supurativa en 50 lactantes y niños. Todos los pacientes del estudio tenían fiebre mayor de 38°C de al menos 6 días duración, a pesar de recibir antibióticos; en 98% de ellos se constató inyección conjuntival sin secreción, signo que hoy se considera como el pilar del diagnóstico clínico de la enfermedad; 86% presentó rash o eritema palmoplantar sin vesículas ni bulas; 96% tenía labios rojos, secos, erosionados y figurados, enantema sin vesículas ni aftas y, en forma ocasional, lengua de “fresa”, lo que liga a la enfermedad con la posibilidad etiológica de Streptococcus pyogenes; 66% de los pacientes desarrolló linfadenitis cervical aguda no supurativa; 44%, edema de manos y pies en lactantes y preescolares; y 98% tuvo acrodescamación, que a partir de la segunda semana, pasada la etapa aguda de la enfermedad, se limitó sólo a manos y pies. El rango de edad de los pacientes fue de 2 meses a 9 años y 1 mes; 54% eran menores de dos años. El síndrome curó espontáneamente, sin secuelas, recurrencias ni contagiosidad (1).
Epidemiología
La incidencia de EK en Japón ha aumentado en forma progresiva desde 1970, posiblemente debido a mejor registro de la patología, llegando a 187 por 100.000 niños menores de 4 años en 2007 y 132 por 100.000 en menores de 5 años. Esta cifra es muy superior a la de Estados Unidos y Europa, donde la incidencia es 3 a 10 por 100.000 niños menores de 5 años. La mortalidad oscila entre 0,1 y 2%. La EK es más frecuente en varones, con una relación de 1,6:1 y 90% de los casos ocurre en menores de 10 años, aunque es poco frecuente en los menores de 6 meses en quienes la presentación clínica es incompleta (Kawasaki atípico) y tiene peor pronóstico. Los niños mayores de 8 años tienen mayor riesgo de desarrollar cardiopatía (2).
Marian Melish fue la primera en describir la enfermedad fuera de Japón, específicamente en Hawaii, en un estudio en que describió número de pacientes y porcentaje acumulativo de casos según edad en niños menores de 18 años (Fig. 1).
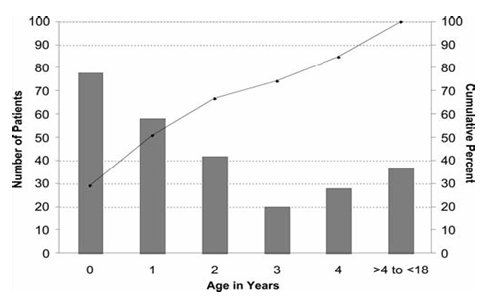 Tamaño completo
Tamaño completo Este estudio demostró la importancia del factor ambiental, ya que la incidencia en niños de origen japonés residentes en Hawaii, quienes poseen un estilo de vida occidental, fue superior a la de los niños japoneses y lo mismo ocurre con niños caucásicos residentes de Hawai, en comparación a aquellos que habitan en el continente (3) (Tabla I).
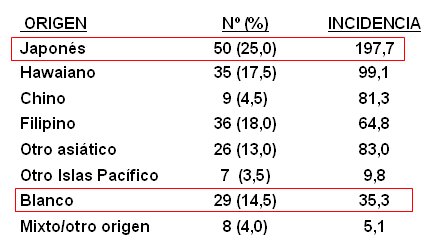 Tamaño completo
Tamaño completo Etiología de la enfermedad de Kawasaki
No está claro si la causa de la EK es un agente único, viral o bacteriano, agentes múltiples con una patogenia común o un superantígeno asociado a un agente; lo que sí está claro es que el resultado final es una marcada activación del sistema inmune.
Trang publicó un estudio en un modelo de ratas en el que se inyectó a estos animales un extracto de pared celular de Lactobacillus casei (EPCLC), después de lo cual se observó la aparición de arteritis y aneurismas coronarios. La respuesta al EPCLC reúne todas las características de la respuesta mediada por superantígenos y lo más relevante fue que la presencia de actividad superantigénica se correlacionó directamente con la inducción de arteritis coronaria (4).
Rowley investigó el origen de la enfermedad en un análisis de varios estudios que describen la presencia de cuerpos de inclusión citoplasmática asociados a múltiples agentes que desencadenan distintas patogenias (5). Para la mayor parte de estos agentes no existe evidencia adecuada o bien, falta confirmación. Actualmente está en investigación la hipótesis de la existencia de un agente viral ARN persistente, previamente no reconocido, que infecta a las células y desata la respuesta inmune induciendo la formación de cuerpos de inclusión citoplasmática persistentes (Tabla II).
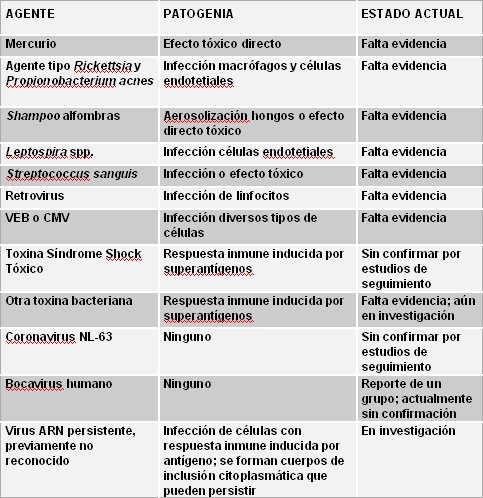 Tamaño completo
Tamaño completo Fisiopatología de la enfermedad de Kawasaki
La formación de aneurismas se explicaría por el paso del agente desde el espacio intravascular al extravascular en el interior de un monocito o macrófago, dando inicio a un fenómeno inflamatorio con agregación plaquetaria y liberación de metaloproteinasas de matriz que alteran la lámina elástica interna y externa, lo que provoca la desestructuración de la pared vascular con formación posterior del aneurisma (Fig. 2).
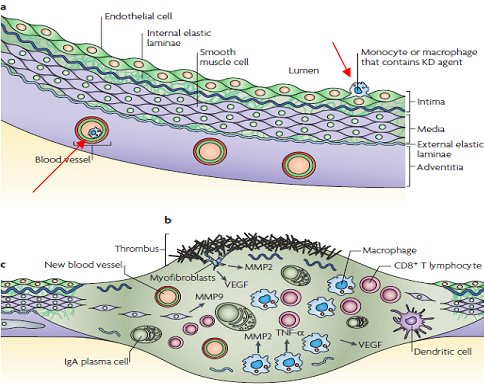 Tamaño completo
Tamaño completo La hipótesis sobre la patogenia de la EK postula que el agente ingresa por la vía respiratoria y penetra a través del epitelio bronquial, donde es captado por los macrófagos tisulares lo que inicia la respuesta inmune innata. Posteriormente el antígeno se transporta hacia los nódulos linfáticos locales, donde desencadenan la respuesta inmune adaptativa. Además estos macrófagos pasan al sistema circulatorio y se dirigen a través de los vasos sanguíneos hacia distintos órganos como páncreas, glándulas salivales, próstata y otros. En el epitelio bronquial el agente desencadena la producción de proteínas virales que se engloban dentro de cuerpos de inclusión citoplasmática que no son reconocidos por el sistema inmune, por lo que pueden permanecer en forma persistente (Fig. 3).
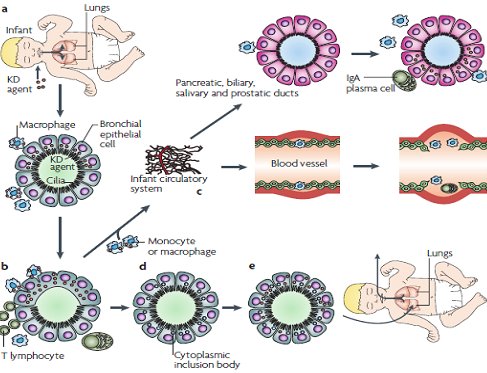 Tamaño completo
Tamaño completo La patología de la EK se caracteriza por un compromiso de las arterias medianas, con inflamación de la túnica íntima y de la zona perivascular que cursa en seis etapas, cuyas características se describen a continuación.
- Etapa I: Desarrollo de edema, proliferación y degeneración del endotelio con descamación y edema del espacio subendotelial; adherencia de fibrina y plaquetas, e inflamación moderada de endotelio, subendotelio y zona perivascular.
- Etapa II: Infiltración del subendotelio por células inflamatorias con desarrollo de edema y desestructuración de la lámina elástica interna. El edema intenso conduce a necrosis de la musculatura lisa media que coexiste con la inflamación de la adventicia.
- Etapa III: Panarteritis necrosante, con infiltración celular inflamatoria de todas las capas; degeneración y necrosis de la pared vascular con desestructuración o fragmentación de las láminas elásticas interna y externa.
- Etapa IV: Proliferación celular de miocitos y fibroblastos en íntima y media; infiltración de células inflamatorias y acumulación fibrinoide en lumen.
- Etapa V: Fibrosis de la íntima y media; en adventicia proliferan las fibras de elastina y colágeno; la pared se engruesa y en casos severos ocurre oclusión o estenosis parcial del lumen.
- Etapa VI: Las láminas elásticas interna y externa se estrechan y fragmentan lo que induce la formación del aneurisma. Las capas íntima, media y adventicia son indistinguibles y se pueden formar trombos en el lumen.
La disociación de la capa media debido al edema ocurre entre 6 y 8 días después del inicio de la enfermedad; el compromiso completo de la pared ocurre a los 10 días; el daño severo y el aneurisma se presentan a los 12 días de evolución, la inflamación persiste hasta los 25 días y las células inflamatorias desaparecen alrededor de los 40 días.
Los órganos afectados por la enfermedad y la correspondiente alteración de éstos se describe a continuación:
- Corazón: Endocarditis, miocarditis y/o pericarditis.
- Tracto digestivo: Glositis, inflamación de glándulas salivales, enteritis, adenitis, colangitis, pancreatitis.
- Respiratorio: Inflamación peribronquial, necrosis y descamación de epitelio bronquial, neumonía intersticial segmentaria, pleuritis y ocasionalmente nódulos pulmonares.
- Tracto urinario: Inflamación intersticial periglomerular, prostatitis y/o cistitis.
- SNC: Meningitis aséptica.
- Sistema linfático: Adenitis con hiperplasia reactiva y/o congestión esplénica.
- La inflamación pulmonar, esplénica, linfática y de glándulas salivales puede ser persistente o recurrente.
Se ha afirmado que la EK es la poliarteritis nodosa infantil (PNI); sin embargo los estudios anatomopatológicos han demostrado que son entidades distintas, porque el compromiso de la EK ocurre antes de que los vasos ingresen a los órganos, tiene una evolución sincrónica con las etapas de la enfermedad y tiene predominio de macrófagos y monocitos en la fase aguda, además de edema; en cambio la PNI ocurre antes y después del ingreso vascular a los órganos, tiene una evolución clínica asincrónica y en la histología predominan los neutrófilos y la necrosis fibrinoide (6).
Diagnóstico de enfermedad de Kawasaki
El patrón clínico consistente con EK incluye fiebre alta con alzas bruscas que dura más de 5 días; desarrollo de eritema y edema palmoplantar, con descamación del lecho ungueal de dedos y ortejos a partir de la segunda a tercera semana desde el inicio; inyección no purulenta de la conjuntiva bulbar; exantema maculopapular escarlatiniforme o multiforme precoz y de duración breve, menor de 5 días; compromiso perianal con descamación; presencia de fisura labial, eritema de la mucosa oral y lengua con aspecto de “fresa”; adenopatía cervical unilateral de más de 1,5 cm de diámetro; y otras manifestaciones como irritabilidad extrema, síntomas cardiovasculares, artritis, artralgias, diarrea, disfunción hepática, meningitis aséptica, leucocituria y uretritis.
No son signos compatibles con EK: fiebre moderada o baja con resolución espontánea en menos de 5 días; descamación palmoplantar precoz; conjuntivitis purulenta; exantema tardío o de más de 5 días de evolución; faringitis sin otras alteraciones de la cavidad oral; adenopatías difusas y paciente en buen estado general. En presencia de estos signos el diagnóstico de EK es dudoso.
Otras pistas clínicas que apoyan el diagnóstico son: eritema e induración de la cicatriz de BCG, rash o descamación perineal, hidrops vesicular, uveítis, hipertensión, meningitis aséptica, poliartritis, enfermedad vascular periférica manifestada como pulso alterado o isquemia y gangrena de extremidades. Asimismo se puede encontrar aumento de PCR y VHS, anemia moderada, leucocitosis y desviación a izquierda, trombocitosis, piuria aséptica, hiponatremia, aumento de transaminasas y gama glutamiltransferasa, hipoalbuminemia, alteración de lípidos plasmáticos, hipereosinofilia más IgE sérica aumentada e hiperrefringencia de las coronarias.
Enfermedad de Kawasaki incompleta
La EK incompleta es una variante de la enfermedad en la cual el paciente no reúne todos los criterios clínicos, por lo tanto se trata de un caso incompleto o atípico que a menudo se diagnostica en forma tardía. Esta forma es más frecuente en el primer año de vida, especialmente en menores de seis meses y los lactantes son más propensos a desarrollar aneurismas coronarios; de hecho la incidencia de aneurismas coronarios en pacientes que no reúnen los criterios diagnósticos alcanza a 25%. Se debe considerar el diagnóstico en todo niño con fiebre de más de cinco días de evolución sin origen explicable, más la presencia de algún criterio adicional. Por ello es fundamental solicitar ecocardiografía en casos de fiebre inexplicable y evidencias de inflamación en los exámenes de laboratorio (VHS y PCR). Según la investigación realizada por Anderson el retardo en el diagnóstico no se asocia con el tipo de profesional médico que atiende al paciente, el número de antimicrobianos utilizados o el número de visitas. Los pacientes con diagnóstico tardío presentan los signos clásicos, pero de comienzo disperso en el tiempo y desarrollan mayor cantidad de aneurismas que los que tienen diagnóstico precoz; por ello es necesario educar para sospechar la enfermedad en niños con fiebre y/o rash (7).
Tratamiento
Newburger estableció en 1986 la efectividad de la inmunoglobulina intravenosa (IgIV) en un estudio controlado, aleatorio y multicéntrico en el cual administró IgIV en dosis de 400 mg/kg/día por un período de cuatro días, asociada a Aspirina® en dosis de 80 a 100 mg/kg/día durante 14 días, a un grupo de pacientes; y sólo Aspirina® a otro grupo. En ambos casos se inició el tratamiento antes del décimo día desde el comienzo de la fiebre. En el grupo que se trató sólo con Aspirina® 28% de los pacientes desarrolló anomalías coronarias, dilatación y aneurismas; en cambio, los pacientes que recibieron IgIV presentaron estas anomalías en sólo 8% de los casos (8).
En 1991 el mismo autor determinó que el mejor efecto se obtiene con una dosis única de 2g/kg de IgIV, que es la indicación que prevalece en la actualidad, gracias a un estudio en que comparó esta dosis con una dosis más baja de 400mg/kg/día por un período de cuatro días, además de administración de Aspirina® en ambos grupos. La incidencia de anomalías coronarias fue inferior con la dosis única (4,6% de los casos) que con la dosis reiterada de 400 mg (9,1% de los casos). Además los marcadores de inflamación, como alfa-1 antitripsina y proteína C reactiva redujeron su valor en forma más precoz con la dosis única (9).
No se conoce con exactitud el mecanismo de acción de la IgIV, pero se recomienda administrar este tratamiento entre el quinto y el décimo día de evolución aguda, puesto que antes de este plazo no previene alteraciones coronarias. Por otra parte se recomienda emplear tal esquema después del décimo día en niños con evidencia de inflamación sistémica, fiebre persistente y aneurismas coronarios. Alrededor de 5% de los niños desarrollan aneurismas a pesar del tratamiento y otro 1% presenta aneurismas gigantes. Respecto a la Aspirina®, se sabe que el uso de ésta en dosis altas, 80 a 100 mg/kg/día junto a la IgIV tiene un efecto antiinflamatorio aditivo. El esquema se mantiene hasta que el paciente permanezca afebril durante 48 a 72 horas y posteriormente se utilizan dosis bajas hasta 6 a 8 semanas tras el inicio de la enfermedad o bien, en forma indefinida en caso de que existan alteraciones coronarias. La Aspirina® no previene el desarrollo de aneurismas aunque se utilice en dosis elevadas.
Se habla de fracaso del tratamiento cuando la fiebre persiste por 36 horas o más tras la administración de IgIV. La incidencia de fracaso de este tratamiento es 10%.
La recomendación actual es aplicar una segunda dosis de Ig de 2 g/kg antes de 10 días de evolución aunque exista respuesta a la dosis inicial, porque además de inducir respuesta en los casos de fracaso al tratamiento inicial esta medida reduce la prevalencia de lesiones coronarias (10). En caso de aneurismas gigantes en fase aguda o subaguda se puede agregar al tratamiento descrito Abciximab, un inhibidor del receptor de la glicoproteína plaquetaria IIb/IIIa, ya que esta medida mejora el pronóstico a largo plazo debido a que disminuye el diámetro de los aneurismas. La adición de metilprednisolona en dosis de 30 mg/kg por vía endovenosa a la terapia con IgIV y Aspirina® dentro de los primeros 9 días de evolución reduce el tiempo de hospitalización y los parámetros inflamatorios, sin embargo no presenta diferencias significativas en la reducción de anomalías coronarias y de todos modos se requiere una segunda dosis de IgIV. Newburger, en un estudio aleatorio, doble ciego y controlado en el que participaron ocho centros con un total de 195 pacientes con EK, concluyó que no hay evidencia suficiente que apoye el uso de pulsos de metilprednisolona junto al tratamiento primario convencional con IgIV (11).
Evolución de la enfermedad de Kawasaki
La EK es una enfermedad autolimitada que cursa con inflamación del sistema vascular que normalmente se resuelve en dos a tres meses. La mayoría de los pacientes están sanos antes de la enfermedad y se recuperan en forma permanente desde el punto de vista funcional, sin secuelas aparentes. El pronóstico a largo plazo (20 a 30 años) depende del estado coronario uno o dos meses desde el comienzo de la enfermedad. La proporción de niños que se recuperan tras recibir el tratamiento con IgIV es superior a 95%; la mayoría tienen un estado inmunológico normal y sólo manifiestan problemas relacionados con el daño coronario.
El compromiso coronario presenta un espectro que varía desde la sobrevida hasta la muerte y puede presentar diversas manifestaciones: ausencia de alteraciones en la ecocardiografía; dilatación coronaria de corta evolución que se resuelve en 2 a 6 meses de evolución; desarrollo de aneurismas medianos, de menos de 7 mm de diámetro; casos con aneurismas gigantes, que miden más de 8 mm; y por último, en menor proporción, muerte precoz durante el curso de la enfermedad.
Recomendaciones de manejo
Los pacientes sin alteración en las ecocardiografías no tienen mayor riesgo de muerte o problemas cardíacos en los siguientes 20 a 30 años, aunque se demuestre la presencia de cambios microscópicos. Por tal razón la recomendación es suspender la dosis bajas de Aspirina® a los dos o tres meses, cuando los parámetros inflamatorios (VHS, PCR y plaquetas) se normalicen; y educar en estilos de vida saludable con énfasis en la prevención del daño cardiovascular o aterosclerosis. Con esto no es necesario hacer control cardiológico entre los dos meses y el año de edad. En los mayores de dos años es necesario controlar con perfil lipídico.
Los pacientes con dilatación coronaria inicial que revierte en 2 a 6 meses tampoco tienen mayor tasa de mortalidad en los siguientes 20 a 30 años; en ella se recomienda administrar dosis bajas de Aspirina® desde el año de edad en forma permanente, además de hacer énfasis en la vida saludable y el control cardiovascular. Se debe repetir la ecocardiografía entre los seis meses y el año de evolución hasta obtener al menos dos ecocardiografías normales. El control cardiológico es opcional.
Los aneurismas medianos se resuelven dentro de los primeros dos años en 50% de los casos, pero las áreas afectadas son anormales, no se dilatan con el ejercicio ni progresan a estenosis en los 20 años siguientes, lo que otorga a la patología un diagnóstico reservado. En 50% de los casos estas alteraciones persisten lo cual depende en parte de su localización, ya que son más frecuentes en determinadas arterias coronarias, formas y tamaños; en este caso pueden progresar a estenosis. Por lo tanto, en caso de aneurismas medianos se recomienda la administración de dosis bajas de Aspirina® en forma indefinida, asociada a drogas antiplaquetarias como clopidogrel o persantina, además de educación en estilo de vida saludable y control cardiovascular.
Los aneurismas gigantes se asocian a riesgo de infarto durante los primeros dos años de evolución. El riesgo de estenosis depende del tamaño, forma y localización del aneurisma, no obstante el riesgo de ruptura aneurismática es bajo y parece estar limitado a los primeros dos años. Por lo tanto, en esta patología se debe aplicar una batería de estudios que incluye ecocardiografía, ECG, holter de arritmias, angiografía, test de perfusión con tecnecio, resonancia magnética, tomografía ultrarrápida y ultrasonido intravascular. El manejo consiste en mantener una adecuada anticoagulación con warfarina más un antiagregante plaquetario, sea Aspirina® o clopidogrel. Se debe realizar estudios imagenológicos en forma periódica con test de ejercicio y drogas, además de SPECT. Se debe efectuar control cardiológico regular y enfatizar en estilo de vida saludable. Se podría requerir tratamientos intervencionales, como plasminógeno tisular, angioplastía con balón en caso de estenosis secundaria de las coronarias, instalación de stent, técnicas de ablación de trombos con rototblader, bypass coronarios en casos de estenosis severa y trasplante cardíaco.
Pronóstico
El pronóstico de la EK ha mejorado en los últimos diez años en forma concomitante con la expectativa de vida, gracias al apoyo cardiológico y a la mayor disponibilidad de tratamientos. Es importante derivar a estos niños a profesionales expertos y mantener una conexión estrecha con ellos; aquellos con problemas graves e inusuales deben ser derivados rápidamente.
Conclusiones
- La etiología de la EK aún no está clara, después de 40 años de la primera descripción.
- La investigación acerca de esta enfermedad ha estado restringida porque se ha supuesto que la EK y la PAN son la misma entidad.
- La población a investigar se ha establecido según la definición de caso, de modo que los estudios se han hecho en aquellos niños que reúnen las condiciones de diagnóstico clásico de EK, dejando fuera a las variantes incompletas o atípicas y por lo tanto, a muchos casos de pacientes con aneurismas.
- Kawasaki planteaba: si la EK es una enfermedad benigna, autolimitada y sin anomalías coronarias, entonces ¿qué es aquella otra entidad con compromiso coronario?
- El riesgo de anomalías coronarias debe ser la base del diagnóstico clínico en aquellos casos que no cumplen con todos los criterios.
- La etiología es el foco de investigación.
- En Chile falta la notificación obligatoria.
