Cursos
← vista completaPublicado el 1 de noviembre de 2004 | http://doi.org/10.5867/medwave.2004.10.3222
Síndrome de arteritis de células gigantes–polimialgia reumática
Giant cell arteritis and polymyalgia rheumatic syndrome
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Simposio Problemas Reumatológicos del Adulto Mayor, organizado en Santiago por la Sociedad Chilena de Reumatología los días 28 y 29 de mayo de 2004.
Presidente: Dr. Carlos Fuentealba. Secretario Ejecutivo: Dr. Pedro Miranda.
La arteritis de células gigantes (ACG) y a la polimialgia reumática (PMR) deben ser consideradas como un síndrome en el que se comparten mecanismos patogénicos y epidemiológicos, pero que puede, presentarse con diferentes expresiones clínicas.
En este síndrome lo característico es el compromiso inflamatorio de la pared de grandes vasos, especialmente ramas extracraneales de la cabeza, ramas de segundo a quinto orden de la aorta y, menos frecuentemente, la aorta misma, en pacientes de edad superior a 50 años. En la PMR el compromiso vasculítico es incompleto y subclínico.
Existen cuatro manifestaciones clínicas de este síndrome que son: Arteritis craneal, PMR aislada, compromiso de grandes vasos ramas de la aorta y/o aortitis, y síndrome inflamatorio sistémico, pudiendo haber mezclas entre las diferentes presentaciones, especialmente con PMR.
Patogenia
Los progresos en la comprensión de la ACG derivan de tres observaciones claves:
- La ACG es una enfermedad dependiente de LT CD4, célula que “organiza” el daño, cuya activación en la pared arterial requiere de la activación de una célula presentadora de antígeno especializada, la célula dendrítica (CD). Las arterias de mediano tamaño, como la arteria temporal, contienen CD residentes que se ubican en el borde limitante con la capa media y que funcionan articulando el sistema inmune innato con el adaptativo.
- En la arteria normal, las CD están inactivas y expresan receptores S-100 y CCR6. Se especializan en captar antígenos y son sensoras del daño celular. En las arterias con vasculitis, las CD se activan expresando CD83 y CD86 y aportan señales coestimuladoras para la activación de LT a través de la IL-18, regulando la liberación de interferón gamma (INF-gama) desde las células T. Además, las CD, en la arteria inflamada, producen grandes cantidades de quemoquinas CCL19 y CCL21 que se ligan a CCR7. La expresión de CCR7 deja atrapadas a las CD activadas y da inicio una respuesta T anómala.
- Existe una “especialidad de sitio” en que las células residentes en la pared arterial responden al ataque inmune en forma diferente según el lugar en que se encuentren.
En cuanto a los mecanismos de daño, en la ACG existen al menos dos diferentes procesos inmunopatogénicos que actúan en forma independiente. El componente vascular, en el que las células inflamatorias infiltran la pared arterial causando un daño estructural que puede llevar a oclusión de su lumen, y el componente inflamatorio sistémico.
Ambos componentes responden a un mismo estímulo, pero parecen tener un mecanismo patogénico diferente. La inflamación vascular se produce por una respuesta inmune adaptativa inadecuada y la inflamación sistémica es secundaria a una excesiva activación del sistema inmune innato.
Componente vascular
La lesión vascular de la ACG depende particularmente de las células T y posiblemente sea gatillada por un antígeno microbiano como virus parainfluenza tipo I, Parvovirus B19, Herpes virus, Clamidia o Micoplasma, aunque faltan evidencias definitivas para asegurarlo.
Los principales componentes del infiltrado de la pared de los vasos son linfocitos T CD4 y macrófagos activados, que parecen entrar a la pared de arterial a través de los vasa vasorum, produciendo un daño inicial en la adventicia, para después penetrar a través de todas las capas de la pared de la arteria.
En la adventicia, las células T encuentran señales estimulatorias, se expanden en forma clonal y liberan citoquinas, especialmente INF-gama, que tiene como blanco principal a los macrófagos. Los macrófagos estimulados por INF-gama son reclutados en la pared del vaso, generando una reacción granulomatosa que produce daño por diferentes vías, como se puede ver en la figura 1.
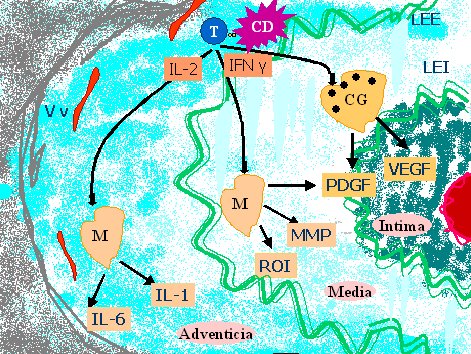 Tamaño completo
Tamaño completo Figura 1. Mecanismo patogénico en el daño tisular de la arteritis de células gigantes.
T= Linfocito T, CD= Célula dendrítica, CG= Célula gigante, M= Macrófago, IL-1= Interleuquina 1, IL-2= Interleuquina 2, IL-6= Interleuquina 6, IFN y= Interferón gamma, ROI= Reactivos intermediarios del oxígeno, MMP= Matriz metaloproteinasa, PDGF= Factor de crecimientoderivado de plaquetas, VEGF= Factor de crecimiento vascular endotelial, Vv= vasa vasorum.
Hay dos factores que determinan el curso de la inflamación en la pared; uno es el tipo de diferenciación de las células T y el otro es la composición celular y de la matriz de la pared del vaso afectado.
En cuanto a la influencia del tipo de diferenciación de células T, si la diferenciación es hacia células T productoras de INF-gama, se correlaciona con una marcada hiperplasia de la íntima y oclusión arterial; si la diferenciación es hacia clones T productores de altos niveles de IL-2 y bajos niveles de INF-gama, la vasculitis evoluciona sin oclusión luminal. La máxima expresión clínica de este mecanismo patogénico es la PMR sin vasculitis por criterios histopatológicos, pero con infiltración de la pared arterial con células T y macrófagos y detección in situ de IL-2, IL-6 e IL-1.
Con respecto a la influencia de la región del vaso afectado, el macrófago activado por estímulos de los LT induce diferentes mecanismos efectores, dependiendo de la región de la pared arterial donde se encuentre:
- En la adventicia se especializan en producir citoquinas proinflamatorias que optimizan la estimulación del LT.
- En la capa muscular lisa de la túnica media la respuesta del macrófago se orienta hacia la producción de reactivos intermediarios del oxígeno (ROI), que inducen peroxidación de lípidos, con daño de la capa muscular, y metaloproteinasas que inducen fragmentación de la lámina elástica interna.
- En la unión media-íntima, los macrófagos liberan factores de crecimiento y factores angiogénicos como Platelet-Derived Growth Factor (PDGF) y Vascular-Endotelial Growth Factor (VEGF).
El ataque inmune activa un programa de respuesta al daño de la arteria, en la cual los miofibroblastos migran y se ubican en el lado endotelial de la íntima, proliferan y depositan matriz extracelular; como resultado de esto, el lumen arterial queda con estenosis u oclusión.
Sin embargo, dado que no siempre se produce obstrucción del lumen, es probable que existan factores de riesgo del huésped que determinen el tipo de reacción arterial frente a la lesión. En todo caso, es importante hacer notar que mientras en las arterias medianas (temporal, subclavia, axilar) se produce obstrucción, en la aorta se producen aneurismas.
Componente inflamatorio sistémico
Esta respuesta se manifiesta como un síndrome inflamatorio sistémico, que está mediado por el sistema inmune innato y representa una respuesta primitiva, caracterizada como una respuesta de fase aguda extrema, donde la IL-6 liberada de los monocitos circulantes juega un rol crítico. Su presencia y grado de activación no son consecuencia de la inflamación vascular, ya que se ha demostrado que los monocitos de los pacientes con PMR sin vasculitis se activan de igual forma (véase figura 2).
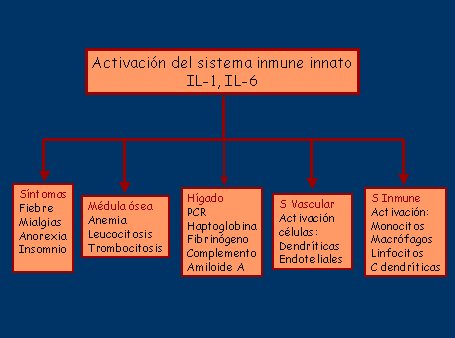 Tamaño completo
Tamaño completo Figura 2. Efectos del sistema inmune innato en el síndrome ACG/PMR.
Epidemiología
El síndrome de ACG y la PMR, así como también los otros cuadros clínicos relacionados, se presentan casi exclusivamente en individuos sobre los 50 años; de hecho, esta edad es uno de los criterios para diferenciar ACG de la Arteritis de Takayasu, que es otra vasculitis de grandes vasos que afecta a la aorta y sus ramas.
La incidencia de ACG y PMR aumenta progresivamente con la edad y es más frecuente en mujeres que en hombres, y la prevalencia de ambas es mayor en países escandinavos y en descendientes de europeos del norte.
La incidencia de ACG es de 15 a 25 casos por 100.000 personas de más de 50 años. Ocurre con mucho menos frecuencia en europeos del sur, que presentan 6 casos por 100.000 y en negros o hispánicos, con 1 a 2 casos por 100.000 individuos de esa edad.
La incidencia de PMR es dos a tres veces más frecuente que la de ACG, con 20 a 25 casos por 100.000 personas sobre 50 años en Europa del norte, y 10 por 100.00 en Europa del sur.
En cuanto a los factores genéticos, se ha asociado al haplotipo HLA-DR4 con un aumento de riesgo de ACG, pero no de la severidad de la enfermedad, a diferencia de lo que pasa con artritis reumatoidea.
Manifestaciones clínicas
El síndrome de ACG y PMR se presenta en forma aguda o insidiosa, en personas sobre los 50 años. Las manifestaciones clínicas son variadas y dependientes del daño vascular o de la inflamación sistémica, como se puede observar en la tabla 1.
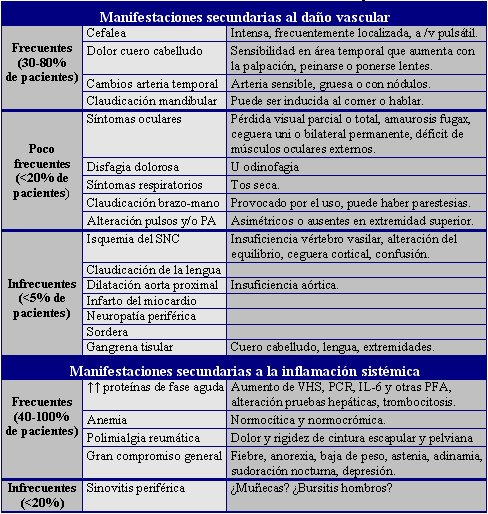 Tamaño completo
Tamaño completo Tabla 1. Manifestaciones clínicas del Síndrome ACG y PMR.
Con respecto a la sensibilidad y especificidad de las manifestaciones clínicas, las manifestaciones comunes, como cefalea o aumento de la VHS, son poco específicas, mientras que la claudicación mandibular o la diplopía son más específicas, pero menos
frecuentes. La sinovitis ha sido asociada con escasa posibilidad de síndrome ACG – PMR.
El clínico debe conocer bien las manifestaciones típicas del síndrome, pero debe estar muy
atento para reconocer las presentaciones atípicas.
Desde el punto de vista clínico se pueden distinguir cuatro presentaciones, que comparten principios patogénicos, no son excluyentes entre sí y pueden tener sobreposición de manifestaciones, como se resume en la figura 3.
- Arteritis craneal (arteritis de la temporal)
- Síndrome de inflamación sistémica.
- Compromiso de la aorta y sus grandes ramas.
- Polimialgia reumática aislada.
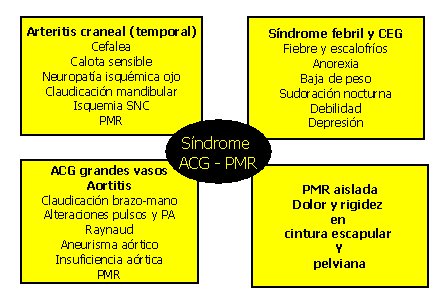 Tamaño completo
Tamaño completo Figura 3. Formas de presentación clínica de la ACG.
Arteritis craneal (arteritis de la arteria temporal)
La mayoría de los pacientes con ACG tienen lesiones en las ramas de las carótidas, como la
arteria temporal superficial, occipital, oftálmica, arteria ciliar posterior y arterias
vertebrales. En 80-90% de los pacientes se encuentran evidencias histológicas de
vasculitis. También pueden comprometerse las carótidas interna y externa.
Clásicamente, consultan por cefalea intensa, muchas veces refractaria al tratamiento con
analgésicos, y sensibilidad en el cuero cabelludo. En 50% de los casos se asocia claudicación de los músculos maséteros y temporales, lo que provoca claudicación
mandibular, síntoma muy específico de la enfermedad. Infrecuentemente se encuentra
isquemia de la lengua, cara, cuello, inflamación de la cara, disfagia u odinofagia.
En el examen, las arterias craneales superficiales, especialmente la temporal, pueden verse
engrosadas o palparse sin pulsos, duras, nodulares y sensibles.
Cuando hay compromiso de las arterias oftálmicas o arterias ciliares se presentan
síntomas visuales, como pérdida visual aguda y dolorosa uni o bilateral, lo que constituye
una emergencia oftalmológica. Cuando se compromete un ojo, existe un 50% de probabilidad de que se afecte también el contralateral si no se trata precozmente.
Otros síntomas oculares son la amaurosis fugax, que consiste en un borramiento visual de corta duración que aparece con el ejercicio o cambios posturales y puede acompañarse de diplopia, visión borrosa postural y diplopia. Puede preceder a la ceguera completa y definitiva. El fondo de ojo puede mostrar edema de papila, hemorragias en astillas y exudados algodonosos en las áreas peripapilares.
Síndrome inflamatorio sistémico con arteritis
La ACG puede presentarse sin hiperplasia de la íntima y sin estenosis ni oclusión del lumen arterial. Estos pacientes no desarrollan isquemia tisular; los síntomas predominantes son los de un síndrome inflamatorio sistémico, que se presenta con fiebre de origen desconocido, pérdida de peso progresiva, sudoración nocturna, anorexia y gran compromiso del estado general.
El riesgo de pérdida visual es menor que cuando hay arteritis craneal. La biopsia de la arteria temporal es el procedimiento diagnóstico de elección y debe ser realizada aunque el examen de la arteria temporal sea normal.
Compromiso de la aorta y sus grandes ramas
Esta forma de presentación está siendo reconocida con más frecuencia que antes, especialmente el compromiso de subclavias y arterias axilares. La biopsia de la arteria temporal muestra vasculitis sólo en 50% de los casos. El diagnóstico se hace con angiografía, no con biopsia.
Las manifestaciones características son el síndrome del arco aórtico, con claudicación de los brazos, ausencia o asimetría de pulsos y presión arterial, parestesias y, a veces, gangrena distal o Reynaud. La inexistencia de síntomas craneales (cefalea, alteraciones visuales o claudicación mandibular) hacen difícil el diagnóstico.
Se estima que el compromiso aórtico ocurre en 10 a 15% de los pacientes, aunque puede ser más frecuente y asintomático. El compromiso craneal habitualmente precede a la aortitis, que puede afectar a cualquier parte de la aorta, pero es más frecuente en el tórax.
A diferencia del compromiso de las arterias craneales, que es obstructivo, en la aorta se produce dilatación o aneurisma. Las consecuencias clínicas incluyen disección o ruptura de la aorta e insuficiencia valvular aórtica.
Desde el punto de vista histológico, la ACG de la aorta puede ser indistinguible de la arteritis de Takayasu. Dado que una ruptura de la aorta puede ser fatal, es importante monitorizar a los pacientes con ACG por varios años después del diagnóstico, mediante radiografía de tórax, TAC o RNM para detectar aortitis.
Polimialgia reumática aislada
El cuadro clínico con que se presenta la PMR consiste en dolor y rigidez en los músculos del cuello, cintura escapular y pelviana, con conservación de los rangos de movimiento pasivo de las articulaciones adyacentes, todo esto acompañado de síntomas sistémicos como fiebre de bajo grado, gran compromiso del estado general, sudoración nocturna y baja de peso.
Aproximadamente 40% de los pacientes con arteritis craneal tienen PMR y 10 a 20% de los con PMR tienen inicialmente vasculitis en la biopsia de la arteria temporal; otras veces, la PMR precede a la arteritis. Cuando los pacientes con arteritis en tratamiento con prednisona recaen, la mayoría de las veces lo hacen con síntomas de PMR.
No se sabe bien en qué sitio se inicia el compromiso inmunológico de la PMR. Se ha dicho que los pacientes con PMR presentan bursitis de hombros y caderas, pero se debe tener mucho cuidado en no confundir PMR con AR del adulto mayor. Se debe considerar que sinovitis es un factor predictor negativo para PMR.
Laboratorio
En el laboratorio, el aumento de la VHS es un elemento muy importante en el diagnóstico del síndrome de ACG y PMR, especialmente con un cuadro clínico compatible, en un paciente de riesgo. Lo mismo sucede con las proteínas de fase aguda, especialmente la PCR, que responde muy directamente a los niveles de IL-6. Como respuesta a la inflamación crónica también se produce anemia normocítica y normocrómica, trombocitosis y alteraciones de las pruebas hepáticas, como elevación de las fosfatasas alcalinas. Hay casos poco frecuentes de ACG y PMR con VHS y PCR normales.
La biopsia para el estudio histológico es fundamental en el diagnóstico de ACG, dado que el laboratorio general es inespecífico y el tratamiento con corticoides no es inocuo. Se debe realizar una biopsia de la arteria temporal, que tiene bajo riesgo, tomando una muestra de tamaño no inferior a 2 a 3 cm. Se debe realizar la biopsia en el lado afectado y si es negativa, pero el cuadro clínico es muy sugerente, en el lado contralateral. Lo ideal es realizar la biopsia antes de iniciar el tratamiento con prednisona, pero no se debe posponer el tratamiento en los pacientes con riesgo de compromiso visual.
Imágenes
El estudio con imágenes se está haciendo cada vez más importante en el síndrome ACG y PMR, especialmente cuando se busca el compromiso de aorta y grandes vasos. Cuando hay obstrucción, ésta se observa con límites progresivos y suaves. Los sitios más afectados son la zona distal de la subclavia, cualquier porción de la arteria axilar y la parte proximal de la arteria braquial. Puede haber compromiso de carótidas y arterias vertebrales, pero no hay compromiso de arterias intracraneales.
Aparte de la angiografía convencional y la angiografía por sustracción digital, el TAC y la RNM son de gran ayuda para observar oclusión o dilatación luminal. Las técnicas de TAC y RNM con medios de contraste son útiles para observar engrosamiento de la pared de los grandes vasos, y la RNM en tres dimensiones, con medio de contraste, sirve para ver alteraciones del arco aórtico.
Tratamiento
El tratamiento de la ACG se basa en los corticoides. Casi todos responden a dosis iniciales de prednisona de 1mg/kg/día, con una respuesta rápida a las 24 - 48 hrs. Esta dosis se mantiene por 4 a 6 semanas, hasta revertir el cuadro clínico y la inflamación sistémica, basándose en la VHS y PCR. Posteriormente se comienza a disminuir muy lentamente, aproximadamente un 10% cada dos semanas.
Con el tratamiento esteroidal ha disminuido la frecuencia de ceguera definitiva y la excelente respuesta a los corticoides se ha considerado como una prueba diagnóstica. El tratamiento debe mantenerse entre 12 a 24 meses y a veces hasta cuatro años, siendo habituales las recaídas.
La PMR puede tratarse con dosis mucho menores de prednisona; habitualmente se inicia con dosis de 20 mg/día, que después de 4 a 6 semanas se disminuyen muy lentamente, aproximadamente 2,5 mg cada dos semanas. También hay frecuentes recaídas y es preciso permanecer expectantes ante un posible paso a ACG craneal o compromiso de grandes vasos.
Los inmunosupresores no han mostrado utilidad en la enfermedad ni han evitado el uso de prednisona. En la actualidad se recomienda agregar al tratamiento Aspirina, en dosis de 100 mg/día y, obviamente, preocuparse de la prevención de la osteopenia u osteoporosis secundaria al tratamiento esteroidal.
El pronóstico de la ACG y PMR es bueno si se trata correctamente. En la ACG las expectativas de vida son las mismas que para la población general, y en la PMR parecen ser aún mayores.
