Reporte de caso
← vista completaPublicado el 1 de julio de 2025 | http://doi.org/10.5867/medwave.2025.06.3050
Caracterización de una variante en el gen KCNH2 en un paciente ecuatoriano con síndrome de QT largo: reporte de caso
Characterization of a variant in the KCNH2 gene in an Ecuadorian patient with long QT syndrome: A case report
Abstract
Long QT syndrome is a rare cardiac channelopathy characterized by prolonged QT intervals and altered T wave morphology. The etiology of long QT syndrome is multifactorial, including environmental and genetic factors. In addition, several heart diseases have been associated with the individual's ethnicity. The objective of the present case report is to describe the genetic and clinical findings of a 44-year-old Ecuadorian man who experienced recurrent episodes of syncope, prolonged QT intervals, and emergent arrhythmias. Through next-generation sequencing, genetic analysis identified a p.Val612Met variant in the KCNH2 gene, associated with long QT syndrome type 2. These findings were key in classifying the patient’s condition as life-threatening and guiding the implementation of a personalized treatment strategy.
Main messages
- Next-generation sequencing allows the identification of genetic variants associated with various conditions.
- Its application is limited by challenges such as restricted accessibility and the requirement for advanced bioinformatics tools for data analysis.
- In the present case report, a p.Val612Met variant was found in the proband, establishing a life-threatening status.
- These results guided the decision of a beta-blocker treatment, resulting in improved symptomatology and clinical management.
Introduction
Long QT syndrome is a rare cardiac channelopathy characterized by prolonged QT intervals and altered T-wave morphology, with an estimated prevalence of 1:2000. Long QT syndrome is associated with an increased risk of sudden cardiac death, syncope, ventricular tachycardia, and fibrillation [1]. The etiology of long QT syndrome is multifactorial, including environmental and genetic factors. In this context, long QT syndrome is predominantly inherited in an autosomal dominant pattern [1].
Furthermore, various cardiac-associated diseases have been linked to an individual’s ethnicity [2,3]. For instance, Hispanic populations exhibit an increased predisposition to carry complex polygenic mutations correlated with cardiac complications [4]. Our research group has published a series of case reports associating cardiac diseases with ancestral components in the Ecuadorian population [2,3].
The objective of the present case report is to describe the genetic and clinical findings of a 44-year-old Ecuadorian man who experienced recurrent episodes of syncope, prolonged QT intervals, and emergent arrhythmias. Using next-generation sequencing, genetic analysis identified a p.Val612Met variant in the KCNH2 gene, associated with long QT syndrome type 2. These findings were key in classifying the patient’s condition as life-threatening and guiding the implementation of a personalized treatment strategy.
Case presentation
This case report was approved by the UTE University Ethics Committee (CEISH-2021-016). Before performing next-generation sequencing, the patient provided written informed consent and consented to including his clinical and imaging data in subsequent publications.
History
A 44-year-old man residing in Quito, Ecuador, presented for medical evaluation due to recurrent episodes of loss of consciousness. These episodes began in his childhood and have been occurring at least once a year, with intermittent symptom-free periods for several years. Recently, he has had two continuous episodes: the first one occurring during the early morning hours, in which the patient described waking up with undefined discomfort in the epigastric and precordial regions, followed by loss of consciousness within seconds. The second episode occurred in the early morning hours, three months later. He awoke with intense distress and chest pain, lost consciousness for approximately 10 seconds, and recovered, feeling extremely fatigued and sweaty.
His personal history included smoking since the age of 18 and occasional alcohol consumption.
Family history reveals an alarming pattern of apparent sudden cardiac deaths: a maternal aunt at age 45, a maternal cousin at age 25, and her daughter died suddenly at age 12 with no prior diagnosis. Additionally, another maternal relative is a carrier of an implantable defibrillator (Figure 1).
Pedigree of the family with long QT syndrome.
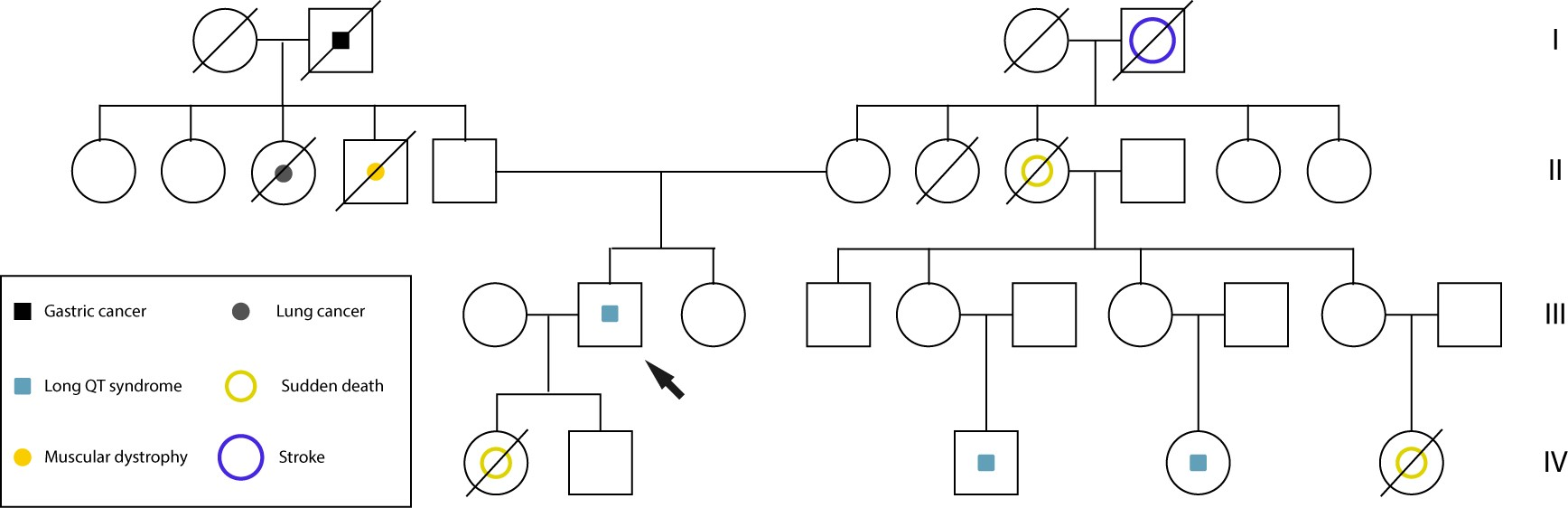
On examination, his echocardiogram did not show any structural cardiac malformations, while his electrocardiogram (ECG) revealed a sinus rhythm with a prolonged QTc interval (579 msec), without specific diagnostic features.
Past medical history
The patient’s past medical history includes a diagnosis of epilepsy at age eight, initially treated with carbamazepine. In addition, the patient suffered a COVID-19-related pneumonia in 2020, which did not require hospitalization.
Molecular and ancestral analysis
A genetic etiology was strongly suspected based on laboratory analysis and the patient’s clinical manifestations. A peripheral blood sample was obtained from the patient for next-generation sequencing to evaluate any potential genetic cause of this patient’s condition. Using the Illumina MiSeq platform and the TruSight Cardio kit, two variants of uncertain significance were identified: c.1834 G>A, p.(Val612Met) in the KCNH2 gene (predicted “probably pathogenic” with 98.5% probability by PolyPhen-2) and c.7142C>A, p.(Ala2381Asp) in the ANK2 gene.
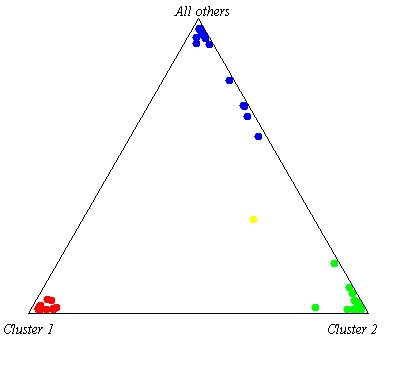
Ancestral analysis using 46 INDEL markers revealed a genetic composition of 50.5% European, 32.2% Native American, and 17.3% African, following the methodology of Zambrano et al. [5] (Figure 2).
Proband’s ancestry composition. The proband is shown in yellow, African ancestry in red, Native American in blue, and European in light green.
Management
Given the recurrent episodes of loss of consciousness, prolonged QT interval on ECG, significant family history of sudden cardiac deaths, and the identification of a likely pathogenic variant in the KCNH2 gene associated with long QT syndrome type 2, a genetic predisposition to life-threatening ventricular arrhythmias is suggested. These findings underscore the need for careful evaluation and treatment.
The patient was advised to discontinue carbamazepine and start taking 20 mg daily of propranolol to reduce the risk of arrhythmias; however, due to the recurring episodes, the dose was increased to 80 mg per day. Regular ECG monitoring will be established to assess changes in the QT interval and detect emergent arrhythmias. Holter monitoring may also be considered for prolonged recording of cardiac activity.
Given the family history, a genetic study in direct relatives is recommended to identify possible carriers of pathogenic variants. Finally, if loss of consciousness persists despite beta-blocker therapy, an implantable cardioverter defibrillator should be considered to mitigate the risk of sudden death. This comprehensive approach aims to control symptoms and improve long-term survival.
Discussion
Long QT syndrome type 2 is a life-threatening, autosomal dominant cardiac disease associated with an increased morbidity and mortality risk. This congenital syndrome is related to mutations in the KCNH2 gene, and its clinical phenotype presents ventricular tachycardia, which could trigger torsade de pointes, syncope, seizures, and sudden death [6].
The long QT syndrome type 2 phenotype depends on the mutation site since variants in the KCNH2 pore domain region are related to a more severe clinical presentation [6]. The KCNH2 gene encodes the α-subunit of the inwardly rectifying potassium channel subunit KV11.1, which generates the rapid activating delayed rectifier potassium current [6].
The most common cause of the inwardly rectifying potassium channel loss of function is its trafficking defect triggered by mutations in the KNCH2 gene. The inwardly rectifying potassium channel pore domain mutations have been related to a dominant-negative effect [6]. This loss-of-function effect is due to the heteromerization between the wild-type and mutant inwardly rectifying potassium channel, which promotes the endoplasmic reticulum's retention and the proteasome system's degradation [7]. Decreased inwardly rectifying potassium channel synthesis, gating defects, and potassium permeation defects are also involved in the loss of function phenotype [6]. All these mechanisms are mainly involved in reducing the rapid activating delayed rectifier potassium current, triggering QTc prolongation, ventricular tachycardia, and sudden death risk [6].
In this case, a pore domain variant p.Val612Met in the KCHN2 gene was detected through next-generation sequencing. The p.Val612Met variant has been previously detected in one individual during a genetic study of 855 unrelated probands. The study reported that carriers with KCNH2 variants showed the presence of QTc prolongation, syncope, or cardiac arrest as the most common clinical features of probands with a KCNH2 variant; however, the clinical data of the carriers were not available [8].
Additionally, a KCNH2 variant (p.Thr613Ala) was detected in a young man who died from sudden cardiac arrest after exercising. In this case, electrophysiological studies revealed a significant reduction in the rapid activating delayed rectifier potassium current compared to the wild-type protein. Furthermore, when co-expressed wild-type and p.Thr613Ala, there was an intermediate phenotype, which suggested no dominant negative effect of the variant [9]. This co-expression effect could help develop pharmacological strategies to inhibit the endoplasmic reticulum retention and proteasome degradation of mutant and wild-type inwardly rectifying potassium channel heterodimers, enhancing their expression in the cellular membrane and improving the rapid activating delayed rectifier potassium current. Moreover, cellular expression and electrophysiological studies of the KCNH2 variant reported here could help to understand the underlying molecular mechanism of loss of function that leads to long QT syndrome type 2 in this proband.
A missense variant in the ANK2 gene (p.Ala2381Asp) was also detected. This gene product (AnkB) regulates the expression and typical localization of Na/K ATPase and Na/Ca exchanger [10]. This variant has not been previously described, although mutations in this gene have been related to ventricular tachycardia [10]. However, electrophysiological studies could also evaluate if the potassium currents are altered when a combination of ANK2 and KCNH2 variants is present. The presence of variants in several proteins involved in ionic currents is related to a more severe phenotype [1]. This severe phenotype could be expressed in the proband’s female relatives.
The proband’s phenotype showed an annual episode of loss of consciousness since he was 8 years old. Genetic tests were recommended based on the recent episodes accompanied by ventricular arrhythmias. The genetic results guided the treatment choice in the patient, and beta-blockers (80 mg daily) were suggested; the patient showed signs of improvement following this decision. Furthermore, periodic controls of ventricular arrhythmias or syncopal episodes are being carried out, mainly due to the sudden death risks based on the proband’s familial history. The lack of genomic tests in female relatives of this proband prevents knowing this hypothesis. However, the use of next-generation sequencing as a method of early identification of risk variants for long QT syndrome type 2 could help the screening of the relatives of affected individuals, to implement an appropriate treatment and prevent sudden death.
Moreover, further research is required to classify the pathogenicity of this variant according to the American College of Medical Genetics and Genomics. Multiple lines of evidence are required to fully assess its impact on cardiovascular diseases, including long QT syndrome. These include case-control and population-based studies to determine the frequency of the variant in affected vs unaffected individuals. Additionally, functional studies using animal models to evaluate its biological implications, and in silico analyses to predict its impact on protein structure and function. In the context of this case report, characterizing pathogenic variants can contribute to the knowledge on genotype-phenotype correlations and guide diagnostic as well as treatment strategies for the patients, ultimately leading to a personalized approach and improving patient care.
The proband’s ancestry showed a high European proportion, which could also affect the susceptibility to long QT syndrome type 2 by KCNH2 mutations [1]. Moreover, the proband’s habits (alcohol consumption and smoking) could also have contributed to the emergence of long QT syndrome symptoms under propranolol treatment. Hence, the effect of these habits on QTc prolongation could also be evaluated in probands harboring ion channel variants.
Conclusions
Identifying the p.Val612Met variant by next-generation sequencing in a proband allowed establishing a life-threatening status. These results guided the decision to use a beta-blocker treatment, which improved symptomatology and clinical management.
