Reporte de caso
← vista completaPublicado el 8 de agosto de 2025 | http://doi.org/10.5867/medwave.2025.07.3051
Secuenciación de Nueva Generación en un paciente Nativo Americano con histiocitos azul marino: Reporte de caso y análisis genómico
Next-Generation sequencing in a Native American patient with sea-blue histiocytosis: A case report and genomic analysis
Abstract
This study presents a case of a 25-year-old Native American woman from Otavalo, Ecuador, diagnosed with sea-blue histiocytosis and myelodysplastic syndrome. Bone marrow aspiration revealed sea-blue histiocytes, and next-generation sequencing identified a likely pathogenic stop-gain mutation in the SAMD9gene, associated with myelodysplastic syndrome. Additionally, variants of uncertain significance were found in the ALB and SRI genes. Ancestral analysis showed a predominantly Native American composition, suggesting a potential genetic predisposition specific to Andean communities. The report underscores the importance of understanding genetic and ancestral backgrounds in diagnosing and managing hematological disorders.
Main messages
- Genomic analysis in a 25-year-old woman with myelodysplastic syndrome identified a pathogenic mutation in the SAMD9 gene and variants of uncertain significance in the ALB and SRI genes. Ancestry analysis showed a predominant Native American component in this individual.
- A key limitation of this case is the need for further research on Native American populations to clarify the role of the identified genetic variants.
- In haematological disorders, it is important to consider genetic factors to support accurate diagnosis and treatment, particularly in specific populations such as Andean communities.
Introduction
Sea-blue histiocytes are macrophages that can accumulate in the spleen, liver, bone marrow, and lymph nodes. They are distinguished by their bright blue color on May-Grünwald Giemsa staining. These cells result from an excessive accumulation of lipids that exceeds the lysosomal capacity of macrophages, leading to incomplete lipid degradation and the accumulation of lipofuscin and ceroid in their cytoplasm [1]. They are commonly associated with thalassemia, lipid metabolism disorders, lysosomal storage diseases, prolonged parenteral nutrition, idiopathic thrombocytopenic purpura, myelodysplastic and myeloproliferative syndromes [2].
Mutations in the SAMD9 gene have been linked to the occurrence of sea-blue histiocytes. Emerging evidence suggests that monosomy 7 —a recurrent cytogenetic abnormality and known etiological factor in myelodysplastic syndromes —may result from the selective loss of chromosome 7, which harbors the SAMD9 locus. Myelodysplastic syndromes represent a group of neoplasms that affect totipotential hematopoietic stem cells, compromising their differentiation, maturation, and morphology in the bone marrow [3].
SAMD9 mutations can be inherited and appear to be more prevalent in specific populations, suggesting a potential genetic predisposition influenced by ancestral heritage. Understanding the interplay between germline mutations and population genetics may offer valuable insights into disease susceptibility, progression, and therapeutic response [4].
In this case, sea-blue histiocytes were identified at the medullary level of a Native American patient through bone marrow aspiration, and a genomic analysis revealed that a mutation in the SAMD9 gene can induce myelodysplastic syndromes associated with sea-blue histiocyte infiltration of the bone marrow.
Case presentation
A 25-year-old woman from Otavalo, Ecuador, with a familial history of anemia and osteoporosis visited a hematology specialist due to epigastric pain, fatigue, migraines, pallor, and a prior diagnosis of gastritis. Physical examination did not reveal any notable findings. However, laboratory results showed significant hematological abnormalities, including a hemoglobin level of 8 g/dL, a platelet count of 115 x 10^3/uL, and a neutrophil count of 1.55 x 10^3/uL. Consequently, she was diagnosed with pancytopenia. Additionally, the mean corpuscular volume and mean corpuscular hemoglobin values were low, in consonance with the diagnosis of anemia. She was treated with 40 mg of oral prednisone daily for eight days.
Due to these findings, the specialist requested a bone marrow aspiration to confirm the diagnosis of myelodysplasia, which could potentially evolve into acute myeloid leukemia. The results of this study revealed increased erythroid series with a myeloid-to-erythroid ratio of 2:1, along with the presence of sea-blue histiocytes (Figure 1).
Bone marrow smear stained with May-Grünwald Giemsa showing sea-blue histiocytes.
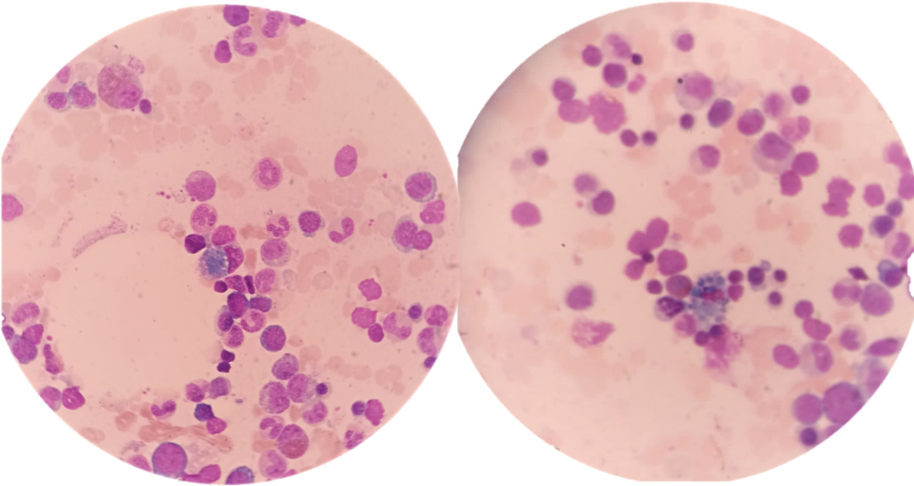
With this diagnosis, the specialist requested an abdominal ultrasound and a lipid profile, which showed normal levels of cholesterol and triglycerides, mild epigastralgia, and inflammatory uropathy. However, no other structural abnormalities were found.
Next Generation Sequencing Analysis
Next-generation sequencing analysis was performed at the Centro de Investigación Genética y Genómica due to the atypical clinical presentation. The individual provided informed consent before the procedure.
Peripheral blood was collected in EDTA, and genomic DNA was extracted using the PureLink Genomic DNA Kit (Invitrogen, USA). Next-generation sequencing was performed with the TruSight One Sequencing Panel (Illumina, USA), following the manufacturer’s instructions. Data analysis was conducted using DRAGEN Enrichment v3.9.5, Annotation Engine v3.15, PolyPhen, SIFT, and Variant Interpreter v2.16.1.300.
Ancestral Component Analysis
To assess the patient’s ancestral composition, a multiplex of 46 ancestry-informative insertion/deletion markers was used. The amplification protocol followed Pereira et al., with a final reaction volume of 10 µL, using Qiagen Multiplex PCR Master Mix (Qiagen), ancestry-informative insertion/deletion marker primers, and genomic DNA [5]. Fragment analysis was performed on a 3500 Genetic Analyzer (Applied Biosystems), with data acquisition using Data Collection v3.3 and analysis in GeneMapper v5. Ancestral composition and population structure were evaluated using STRUCTURE v2.3.4, following the methodology described by Zambrano et al. [6].
Results
The genomic analysis revealed a total of 7563 variants; among them, a stop gain in the SAMD9 gene (p.Trp513Ter) was identified and predicted as likely pathogenic. Additionally, two missense variants of uncertain significance (VUS) were detected: one in the ALB gene (p.Phe533Cys) and another in the SRI gene (p.Asp158Asn) (Table 1).
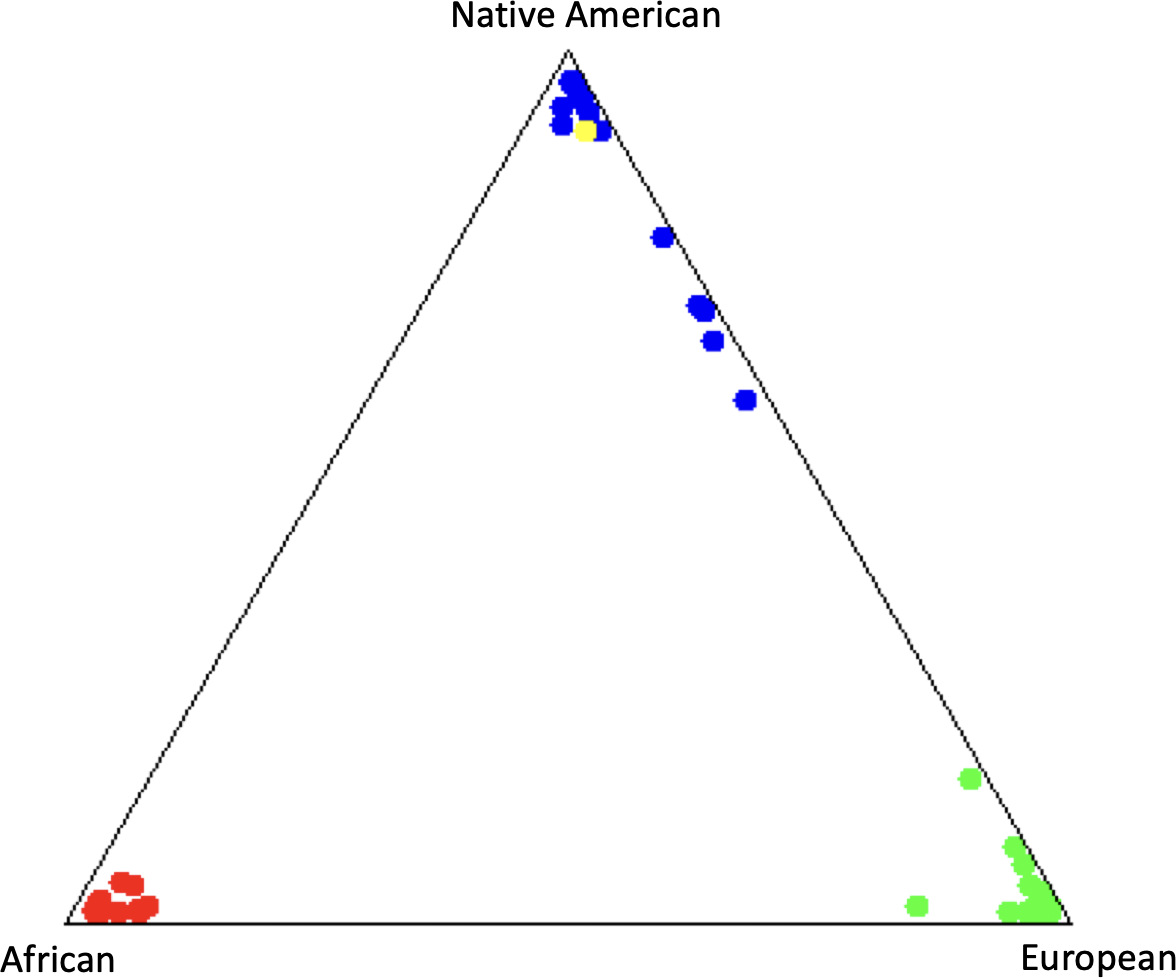
The ancestral component analysis revealed a predominantly Native American composition, with proportions of 94% Native American, 4,4% European, and 1,6% African ancestry (Figure 2).
Ancestral Composition of the subject in yellow.
Discussion
Sea-blue histiocytosis is a rare histological finding associated with lipid metabolism disorders, lysosomal storage diseases, and myelodysplastic and myeloproliferative syndromes [2]. In this case, blue histiocytes were identified through bone marrow aspirates. Furthermore, genetic analysis may contribute to elucidating whether these histiocytes represent a primary manifestation of a genetic syndrome or a secondary phenomenon linked to an underlying hematologic disorder.
Genomic analysis revealed a heterozygous stop-gain variant in the SAMD9 gene, located on chromosome 7q21.2. This gene encodes a protein involved in cell proliferation, apoptosis, and response to cellular stress. The identified variant introduces a premature termination codon, likely resulting in a truncated, non-functional protein. According to the ACMG/AMP guidelines, this variant fulfills the criteria PVS1 and PM2, supporting its classification as likely pathogenic [7]. Pathogenic variants in SAMD9 have been implicated in a spectrum of disorders, including myelodysplastic syndrome, leukemia with the 7-monosomy syndrome, ataxia-pancytopenia syndrome, and MIRAGE syndrome (Myelodysplasia, Infection, Restriction of growth, Adrenal hypoplasia, Genital problems, and Enteropathy) [3].
The loss of SAMD9 protein function could cause dysregulated hematopoiesis and the accumulation of dysfunctional macrophages, which are characteristic of sea-blue histiocytosis. The patient’s sea-blue histiocytes, anemia, and pancytopenia were consistent with this phenotype, indicating myelodysplastic syndrome and a possible progression toward acute myeloid leukemia.
In addition to the SAMD9 variant, two variants of uncertain significance were identified in the ALB and SRI genes. The ALB gene encodes albumin, the most abundant plasma protein, which plays a critical role in maintaining oncotic pressure and transporting endogenous and exogenous molecules. Although 74 nucleotide substitutions in ALB have been reported, none have been definitively linked to disease [8]. In contrast, the variant identified in the Ecuadorian individual is classified as a variant of uncertain significance based on ACMG/AMP criteria, supported by PM2 (absence from population databases) and PP3 (multiple computational lines of evidence suggesting a deleterious effect) (Table 1). These findings may indicate potential functional relevance. The variant identified in this patient is absent from population databases (PM2) and predicted to be deleterious by multiple in silico tools (PP3), suggesting potential functional relevance. A dysfunctional albumin protein could impair lipid transport, potentially contributing to lipid accumulation in macrophages and the development of sea-blue histiocytosis [9].
Moreover, sorcin, a protein that controls intracellular calcium homeostasis and stress response, is encoded by the SRI gene. Specifically, sorcin maintains calcium balance in the endoplasmic reticulum and lysosomes, thereby regulating lipid metabolism and macrophage homeostasis [10]. Conversely, lysosomal accumulation of various substrates, such as proteins and lipids, is linked to calcium dysregulation, which disrupts processes like autophagy and endocytosis, triggering defective lipid degradation and histiocyte accumulation [11]. In the identified variant of variants of uncertain significance in the SRI gene, the substitution of aspartic acid with asparagine at position 158 in sorcin may influence its calcium-binding affinity or protein-protein interactions, which could be associated with alterations in these critical cellular processes.
The pathogenicity of these variants of uncertain significance in the ALB and SRI genes remains unclear, and further clinical testing is required to elucidate their role in hematological disorders. However, the interaction between the likely pathogenic SAMD9 variant, which could trigger myelodysplastic syndrome, may be modulated by variants of uncertain significance present in the ALB (lipid transport) and SRI (endocytic responses) genes. Future research should focus on including functional assays in in vitro models, gene expression analysis in patient-derived cells, among others. In addition, expanding studies in underrepresented and ancestrally diverse populations could clarify their importance.
Additionally, the high Native American ancestral component (94%) suggests that the likely pathogenic variant in the SAMD9 gene, which has not been previously reported, may be specific to the Andean communities of Ecuador. The influence of altitude on the expression of phenotypic mutations in the SAMD9 gene could be related to the alteration of cellular stress and hematopoiesis pathways induced by hypoxia [12].
Understanding a patient’s ancestral background can provide insights into their genetic risk factors for conditions such as myelodysplastic and the presence of sea-blue histiocytes. However, a significant limitation of this study was the inability to perform a follow-up, as the patient did not return for subsequent medical evaluations or respond to further contact attempts. This restricted the opportunity to assess disease progression and the clinical impact of the identified genetic findings. Further studies in Native American populations are necessary to elucidate the role of SAMD9 variants in hematological disorders.
Conclusions
This case report highlights the significance of genetic analysis in diagnosing rare hematological disorders such as sea-blue histiocytosis and myelodysplastic. The identification of a likely pathogenic mutation in the SAMD9 gene, along with variants of uncertain significance in the ALB and SRI genes, underscores the complexity of these conditions and the potential for genetic predisposition influenced by ancestral background. The high Native American ancestry of the patient suggests that certain genetic mutations may be more prevalent in specific populations, emphasizing the need for population-specific genetic studies. Understanding the genetic and ancestral factors can improve diagnostic accuracy, inform clinical management, and guide future research into the pathophysiology and treatment of hematological disorders.
