Reporte de caso
← vista completaPublicado el 20 de junio de 2017 | http://doi.org/10.5867/medwave.2017.05.6978
Enfermedad de neuro-Behçet en Perú: reporte de caso y revisión de la literatura
Neuro-Behçet's disease in Peru: a case report and literature review
Resumen
La enfermedad de Behçet es una vasculitis que puede ocasionar lesiones inflamatorias en múltiples órganos o sistemas como el neurológico. El mayor número de casos a nivel mundial se han reportado a lo largo de la llamada Ruta de la Seda, que va desde la región mediterránea hasta Japón, siendo considerado una enfermedad rara en países latinoamericanos. La frecuencia de afectación neurológica oscila en un rango entre 5 y 13%. Se presenta el caso de una mujer adulta joven con criterios diagnósticos de enfermedad de Behçet y manifestaciones de afectación neurológica, así como una revisión de la literatura en Perú.
Introducción
La enfermedad de Behçet es una vasculitis que ocasiona lesiones inflamatorias en múltiples órganos como la piel, las articulaciones y en los sistemas gastrointestinal, renal, cardiopulmonar y/o neurológico [1].
La mayor cantidad de casos a nivel mundial se han reportado a lo largo de la llamada Ruta de la Seda, que va desde la región mediterránea hasta Japón. Se presenta sobre todo en la tercera década de la vida, afectando más a hombres que mujeres (1,4:1), con mayor incidencia de complicaciones sistémicas graves en los hombres jóvenes [2]. En países de América su incidencia es mucho más baja, llegando a catalogarse como una enfermedad rara [3]. La frecuencia reportada de afectación neurológica, oscila en un rango entre cinco y trece por ciento. Se ha reconocido que esta afectación aumenta la mortalidad y morbilidad de los pacientes con enfermedad de Behçet [4],[5].
El diagnóstico se basa en los siguientes criterios: úlceras orales recidivantes, generalmente dolorosas (como mínimo tres recidivas en un año); junto a dos o más de estos síntomas: úlceras genitales recidivantes, lesiones oculares como hipopion, uveitis, vasculitis retiniana, lesiones cutáneas y prueba de patergia positiva según los criterios propuestos por el Grupo de Estudio Internacional en 1990 (Tabla 1) [6]. Recientemente se han revisado estos criterios por presentar baja sensibilidad. Asimismo, se han establecido nuevos criterios internacionales para la enfermedad de Behçet mediante una escala de 10 puntos, donde un puntaje mayor de 4 confirma el diagnóstico. En esta puntuación se agrega a los criterios anteriores, las manifestaciones neurológicas y manifestaciones vasculares (trombosis arterial, venosa o flevitis). Además, es el test de patergia es un punto adicional que se agregará cuando este sea realizado [7] (Tabla 2).
El propósito del presente artículo es reportar un caso de neuro-Behçet, revisar los casos reportados en Perú hasta la fecha e identificar sus características comunes.
 Tamaño completo
Tamaño completo 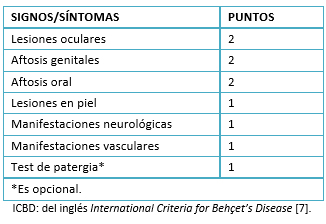 Tamaño completo
Tamaño completo Métodos
El diseño del presente estudio es un reporte de caso de corte retrospectivo. Para su realización se solicitó y contó con el consentimiento informado para publicación, de acuerdo a las recomendaciones éticas internacionales. Se hizo la búsqueda de literatura médica referente a casos reportados de enfermedad de neuro-Behçet en Perú (MeSH: Symptom Complex Triple OR Adamantiades-Behcet Disease OR Behcet Triple Symptom Complex OR Old Silk Route Disease OR Behcet's Syndrome OR Behçet Disease AND Case Reports -Publication Type- AND Neurologic Manifestations) en PubMed, Lilacs y SciELO; identificándose seis casos en el país [7],[8],[9]. Se aplicaron los criterios y el puntaje necesario para el diagnóstico de la enfermedad de Behçet.
Reporte de caso
Mujer de 20 años, procedente de la Provincia Constitucional del Callao, con ascendencia europea-mediterránea por ambos padres. Paciente con antecedentes de lesiones ulcerativas dolorosas en mucosas lingual, paladar duro y labios tres años previos a su diagnóstico con un curso progresivo y remisión espontánea.
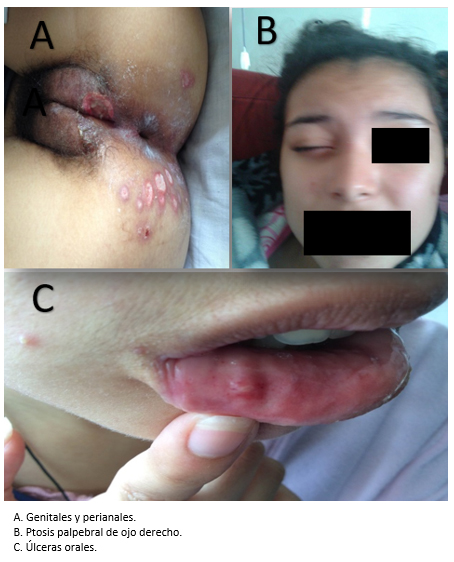
Un año antes del diagnóstico manifestó un episodio de visión borrosa de tres a cuatro días, que mejoró sin tratamiento. Siete meses antes, tuvo un episodio de uveítis anterior en ojo derecho con remisión espontánea después de dos días. Al siguiente mes, presentó recidiva en ojo izquierdo con las mismas características (Figura 1). Cuatro meses antes, cursó con artralgias intermitentes de 8/10 de intensidad que cedían con tratamiento analgésico y antiinflamatorio. Dos meses antes, manifestó temblor posicional en miembros superiores con duración de tres días y remisión espontánea. Un mes antes, tuvo úlceras genitales de remisión espontánea en dos semanas sin dejar cicatriz y con recurrencia hasta el momento del diagnóstico. Durante este tiempo refirió constipación, que mejoró con lactulosa.
Acudió a emergencia tras agregarse, semanas antes y de forma brusca, diplopía vertical que no cedía de forma espontánea.
 Tamaño completo
Tamaño completo La paciente presentó en el examen físico de su admisión un habla fluente, pupilas anisocóricas (derecha: 2,5 centímetros e izquierda: 3,5 centímetros), dificultad para dirigir la mirada hacia abajo con el ojo izquierdo, nistagmus vertical, escotoma central y ptosis palpebral en el ojo derecho. El fondo de ojo evidenció vitreitis, edema macular y focos de periflebitis periférica a predominio del ojo derecho. También se presentaron úlceras dolorosas genitales y perianales de bordes poco definidos de uno a dos centímetros de diámetro (Figura 1).
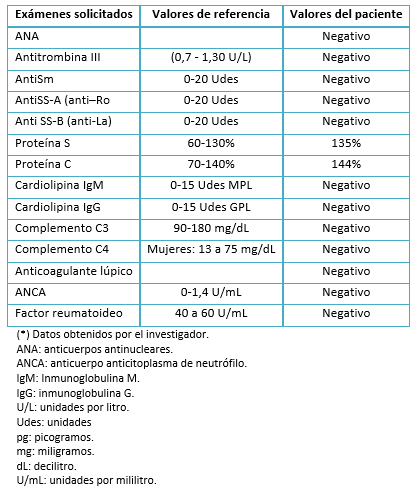
Los anticuerpos antifosfolipídicos, anticuerpos antinucleares, anticuerpos anticitoplasma de neutrófilos, factor reumatoide, complemento sérico y bandas oligoclonales, resultaron negativos (Tabla 3). Por la ligera elevación de proteína C y S y velocidad de sedimentación, se sospechó de encefalopatía postinfecciosa y/o rombencefalitis. Sin embargo, en el estudio de líquido cefalorraquídeo no se encontró indicadores de algún agente etiológico (Tabla 4).
 Tamaño completo
Tamaño completo 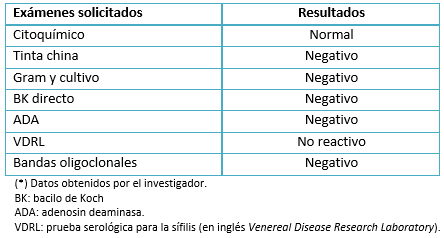 Tamaño completo
Tamaño completo Los resultados de resonancia magnética nuclear muestran imágenes hiperintensas en ponderados FLAIR y T2 en región medial mesencefálica bilateral, con mayor compromiso izquierdo e hipocampo derecho (Figura 2). Los potenciales evocados visuales evidenciaron latencias incrementadas bilaterales a predominio izquierdo.
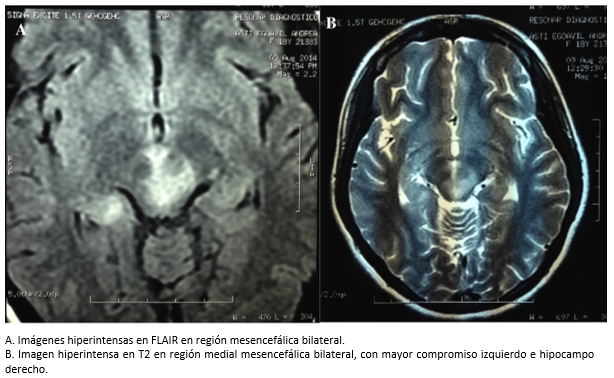 Tamaño completo
Tamaño completo Se determinó que el cuadro clínico correspondía a una enfermedad inflamatoria, sin evidencias de infección. Los síntomas y signos extra neurológicos y las características de las lesiones del sistema nervioso central, permitieron excluir otras causas de lesiones inflamatorias en este sistema como de lupus eritematoso, artritis reumatoide, herpes, VIH, sífilis, esclerosis múltiple, encefalopatía postinfecciosa y rombencefalitis viral.
Las presencia de úlceras orales y genitales sin etiología definida, nos llevó a sospechar de la enfermedad de Behçet. Por este motivo se solicitó la prueba de patergia [7] (Figura 3), que resultó negativa. Sin embargo, en la muestra de biopsia del borde de la úlcera se evidenció infiltrado inflamatorio agudo y crónico, superficial y profundo de disposición perivascular e intersticial. Todas estas lesiones eran compatibles con enfermedad autoinmune (Figura 4).
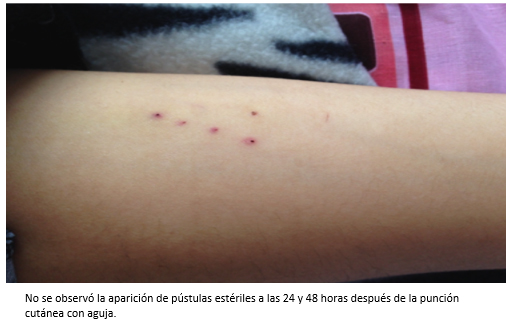 Tamaño completo
Tamaño completo 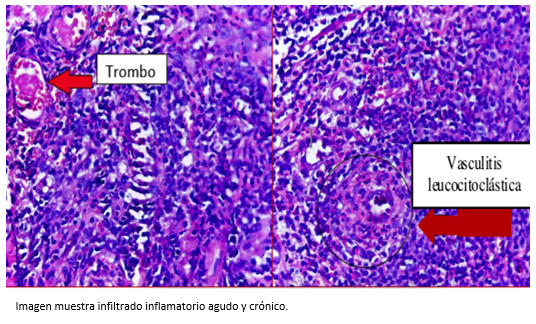 Tamaño completo
Tamaño completo Adicionalmente, el hemograma presentó leucocitosis (14 440 células por milímetros cúbicos) a predominio de linfocitos; hemoglobina en 9,1 gramos por decilitro con saturación de transferrina de 7,75%; ferritina 57 nanogramos por mililitro y hierro de 17,49 microgramos por decilitros (Tabla 4). El examen de orina con leucocitos mayor de 100 por campo, dio positivo al cultivo de Candida albicans al igual que el examen de secreción vaginal (Tabla 5).
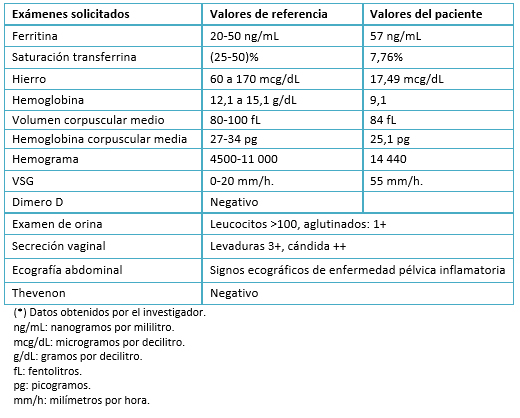 Tamaño completo
Tamaño completo Luego de tratar las infecciones y estabilizar a la paciente, se inició tratamiento con prednisona (a dosis de 0,5 a 1 milígramo por kilogramo de peso al día) que ha continuado hasta la actualidad con descenso progresivo. También recibió tratamiento durante dos meses con azatioprina a dosis de 2 a 2,5 milígramos por kilogramo de peso al día. Durante este período presentó meningoencefalitis de probable etiología bacteriana, por lo que se decidió retirar el tratamiento corticoide e inmunosupresor mientras se manejaba esta complicación. La paciente presentó una evolución caracterizada por infecciones urinarias recurrentes, síndrome de Cushing y osteopenia como reacciones adversas al tratamiento con corticoides. Además, presentó como secuela disminución de agudeza visual a predominio derecho
Diagnóstico según criterios
El cuadro clínico de la paciente cumplió con el criterio mayor (úlceras orales recurrentes) y dos de los cuatro criterios adicionales establecidos por el Grupo de Estudio Internacional en 1990 (úlceras genitales y uveítis anterior como lesión ocular) [1] (Tabla 1). Además, cumplió con los nuevos Criterios Internacionales para la Enfermedad de Behçet establecidos en 2014, al presentar ocho de un puntaje máximo de 10, que les adiciona a los criterios clásicos lesiones en la piel (aftas) y manifestaciones neurológicas sin presentar manifestaciones vasculares como trombosis arterial, venoso o flebitis [7] (Tabla 2).
Casos identificados
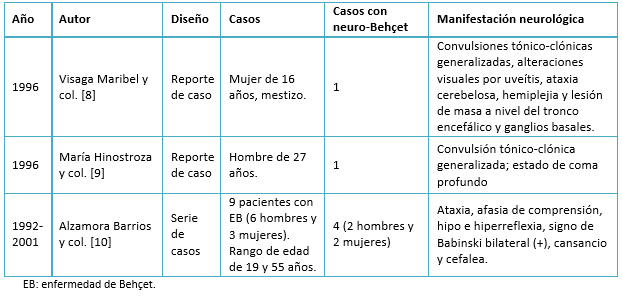
Los reportes de enfermedad de Behçet en el Perú son escasos. Sólo se identificaron mediante la búsqueda 6 casos de neuro-Behçet en los últimos 25 años los que se describen en la Tabla 6.

Autores
Roberto A. Molina
Jirón Libra 1114, Urbanización Mercurio Bajo, Los Olivos, Lima, Perú
Andrely Huerta-Rosario
Carlos Alexander Alva Díaz
Koni Katerin Mejía Rojas
Nicanor Mori
Servicio de Neurología, Departamento de Medicina, Hospital Daniel Alcides Carrión, Callao, Perú
Roberto Romero Sánchez
Figuras
Foro
Comentar
Comentarios
Historial
Citación Molina RA, Huerta-Rosario A, Alva Díaz CA, Mejía Rojas KK, Mori N, Romero Sánchez R. Neuro-Behçet's disease in Peru: a case report and literature review. Medwave 2017;17(05):e6978 doi: 10.5867/medwave.2017.05.6978
Envío 02/02/2017
Aceptación 04/06/2017
Publicación 20/06/2017
Notas
Palabras claves
Behçet's disease, neurological involvement, vasculitis
Origen y arbitraje
no solicitado. con revisión por tres pares revisores externos, a doble ciego
Referencias
- Ideguchi H, Suda A, Takeno M, Ueda A, Ohno S, Ishigatsubo Y. Behçet disease: evolution of clinical manifestations. Medicine (Baltimore). 2011 Mar;90(2):125-32. | CrossRef | PubMed
- Savey L, Resche-Rigon M, Wechsler B, Comarmond C, Piette JC, Cacoub P, et al. Ethnicity and association with disease manifestations and mortality in Behçet's disease. Orphanet J Rare Dis. 2014 Mar 27;9:42. | CrossRef | PubMed
- Castillo-González W, González--Argote J, Hernández-Estévez J. Enfermedad de Behçet. Revista Cubana de Reumatología. 2014;16(3):12. | Link
- Yoon DL, Kim YJ, Koo BS, Kim YG, Lee CK, Yoo B. Neuro-behçet's disease in South Korea: clinical characteristics and treatment response. Int J Rheum Dis. 2014 May;17(4):453-8. | CrossRef | PubMed
- Zouboulis CC, Vaiopoulos G, Marcomichelakis N, Palimeris G, Markidou I, Thouas B, et al. Onset signs, clinical course, prognosis, treatment and outcome of adult patients with Adamantiades-Behçet's disease in Greece. Clin Exp Rheumatol. 2003 Jul-Aug;21(4 Suppl 30):S19-26. | PubMed
- Criteria for diagnosis of Behçet's disease. International Study Group for Behçet's Disease. Lancet. 1990 May 5;335(8697):1078-80. | PubMed
- International Team for the Revision of the International Criteria for Behçet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014 Mar;28(3):338-47. | CrossRef | PubMed
- Visaga M, Campos P, Bancalari E, Romero F, Hernandez H, Berrocal A, Trelles L, Calvo Armando, et al. Neurobehcet pediátrico. Presentación de un caso y revisión de la literatura. Rev Med Hered. 1996;7:178-181. | Link
- Hinostroza M, Dios J. Neuro-Behcet. Rev Soc Peruana Med Interna. 1996; 9(3). | Link
- Alzamora B, Martínez F. Enfermedad de Behcet: estudio clínico y tratamiento en el Hospital Arzobispo Loayza. Rev Med Hered. 2001;12(2): 58-64. | Link
- International Team for the Revision of the International Criteria for Behçet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014 Mar;28(3):338-47. | CrossRef | PubMed
- Cho SB, Cho S, Bang D. New insights in the clinical understanding of Behçet's disease. Yonsei Med J. 2012 Jan;53(1):35-42. | CrossRef | PubMed
- Kalra S, Silman A, Akman-Demir G, Bohlega S, Borhani-Haghighi A, Constantinescu CS, et al. Diagnosis and management of Neuro-Behçet's disease: international consensus recommendations. J Neurol. 2014 Sep;261(9):1662-76. | CrossRef | PubMed
- Aydin MD, Aydin N. A neuro-Behcet's lesion in oculomotor nerve nucleus. Acta Neurol Scand. 2003 Aug;108(2):139-41. | PubMed
- Tugal-Tutkun I, Onal S, Ozyazgan Y, Soylu M, Akman M. Validity and agreement of uveitis experts in interpretation of ocular photographs for diagnosis of Behçet uveitis. Ocul Immunol Inflamm. 2014 Dec;22(6):461-8. | CrossRef | PubMed
- Siva A, Saip S. The spectrum of nervous system involvement in Behçet's syndrome and its differential diagnosis. J Neurol. 2009 Apr;256(4):513-29. | CrossRef | PubMed
- Mat C, Yurdakul S, Uysal S, Gogus F, Ozyazgan Y, Uysal O, et al. A double-blind trial of depot corticosteroids in Behçet's syndrome. Rheumatology (Oxford). 2006 Mar;45(3):348-52. | PubMed
- Saadoun D, Wechsler B, Terrada C, Hajage D, Le Thi Huong D, Resche-Rigon M, et al. Azathioprine in severe uveitis of Behçet's disease. Arthritis Care Res (Hoboken). 2010 Dec;62(12):1733-8. | CrossRef | PubMed
- Whitcup SM, Salvo EC Jr, Nussenblatt RB. Combined cyclosporine and corticosteroid therapy for sight-threatening uveitis in Behçet's disease. Am J Ophthalmol. 1994 Jul 15;118(1):39-45. | PubMed
- Kaklamani VG, Kaklamanis PG. Treatment of Behçet's disease--an update. Semin Arthritis Rheum. 2001 Apr;30(5):299-312. Erratum in: Semin Arthritis Rheum 2001 Aug;31(1):69. | PubMed
- Kalra S, Silman A, Akman-Demir G, Bohlega S, Borhani-Haghighi A, Constantinescu CS, et al. Diagnosis and management of Neuro-Behçet's disease: international consensus recommendations. J Neurol. 2014 Sep;261(9):1662-76. | CrossRef | PubMed
- Hatemi G, Silman A, Bang D, Bodaghi B, Chamberlain AM, Gul A, et al. EULAR recommendations for the management of Behçet disease. Ann Rheum Dis. 2008 Dec;67(12):1656-62. | CrossRef | PubMed
- Saadoun D, Wechsler B, Desseaux K, Le Thi Huong D, Amoura Z, Resche-Rigon M, et al. Mortality in Behçet's disease. Arthritis Rheum. 2010 Sep;62(9):2806-12. | CrossRef | PubMed