Congresos
← vista completaPublicado el 1 de noviembre de 2007 | http://doi.org/10.5867/medwave.2007.10.1258
Pubertad retrasada
Delayed puberty
Resumen
Este texto completo es la transcripción editada y revisada de la conferencia dictada en el marco del V Congreso de Obstetricia, Ginecología Infantil y Adolescencia, realizado en Santiago entre los días 31 de agosto al 2 de septiembre de 2006. El evento fue organizado por la Sociedad Chilena de Obstetricia, Ginecología Infantil y Adolescencia.
Presidente: Dra. Pamela Oyarzún.
Introducción
Hasta agosto de 2005, había en Pubmed alrededor de 3.700 artículos relacionados con alteraciones en el inicio de la pubertad, entre ellos algunos que comunicaban experiencias en animales. 3.100 artículos se referían a estudios en seres humanos y de ellos, 72 trataban sobre pubertad precoz y 28%, sobre pubertad retrasada (Tabla I, arriba). En la búsqueda por sexo, como la pubertad retrasada afecta mayoritariamente al sexo masculino, el porcentaje de artículos sobre el problema en varones fue de 56,1% (Tabla I, abajo).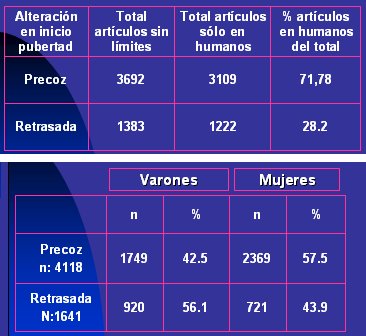 Tamaño completo
Tamaño completo Tabla I. Resultado de búsqueda sobre alteraciones de la pubertad (Pubmed, agosto 2005)
Pubertad retrasada
La pubertad retrasada es la ausencia de caracteres sexuales secundarios, a 2 desviaciones estándar de la edad promedio del inicio de la pubertad normal en la población y sexo a los que pertenece el individuo. En términos prácticos y cronológicos, para los varones es a los 14 años, cuando el volumen testicular es menor de 4 ml y para las mujeres es a los 13 años, cuando aún no hay presencia de telarquia. Una situación especial es la pubertad detenida, que corresponde a un grupo de pacientes que pueden tener anormalidades parciales o transitorias; inician la pubertad a una edad normal, pero transcurren más de cinco años entre el primer signo puberal y el desarrollo gonadal completo en hombres o la aparición de menarquia, en mujeres.La pubertad retrasada es un cuadro clínico frecuente que ocurre en 3% de la población, pero hay pocos estudios, y los que hay no son homogéneos, en el análisis de las diferentes causas de pubertad retrasada; también es más frecuente en varones, pero en ambos sexos la variedad más frecuente es la pubertad retrasada de tipo simple, que por lo general es de origen familiar o idiomática y se debe a retraso constitucional del crecimiento y la pubertad. 60% de los casos se da en varones y 30%, en mujeres.
Las causas más frecuentes de pubertad retrasada son: las enfermedades crónicas, como asma, enfermedad celíaca, enfermedad de Crohn, hipotiroidismo primario, terapia prolongada con glucocorticoides, anorexia nerviosa; y los cuadros fisiológicos que no representan enfermedad, como el retraso constitucional del crecimiento y la pubertad. Otras patologías constituyen causas de pubertad retrasada poco frecuentes, pero es un gran logro conocerlas en detalle, pues su estudio molecular ha permitido entender el complejo fenómeno de la pubertad; entre ellas se pueden mencionar: los trastornos del eje hipófisis-hipotálamo, como el síndrome de Kallmann y las deficiencias congénitas o adquiridas de hormonas hipofisiarias; síndromes dismórficos, como los síndromes de Noonan y de Prader-Willi; anomalías en los cromosomas sexuales, como los síndromes de Turner y Klinefelter); y otras causas de insuficiencia gonadal, como agénesis-disgénesis gonadal, cirugía, radioterapia, quimioterapia, insuficiencia ovárica primaria, insuficiencia gonadal autoinmune y galactosemia, en las niñas. En la Tabla II se resumen las causas de pubertad retrasada y detenida.
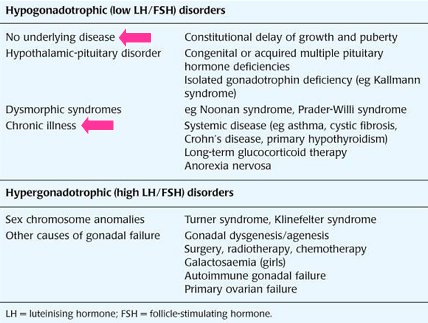 Tamaño completo
Tamaño completo Tabla II. Principales causas de pubertad retrasada y detenida
Genética de la pubertad retrasada
El genotipo en las alteraciones del eje HHG se resume de la siguiente manera:- Hipotálamo: GnRH/migración; defectos en la síntesis y liberación de GnRH; alteraciones de leptina y su receptor; alteraciones de factores de transcripción.
- Hipófisis: Mutaciones en el receptor de GnRH; alteraciones en el desarrollo; alteraciones en producción hormonal
- Gónadas: Alteraciones en las subunidades de FSH y LH; mutaciones en los receptores FSH y LH; alteraciones en las señales intracelulares; alteraciones en la función y diferenciación.
Para entender las causas de la pubertad retrasada se debe conocer las bases génicas, especialmente los siguientes genes:
- De la migración de las neuronas que producen hormona liberadora de gonadotrofinas (GnRH): gen KAL
- De la acción del GnRH: gen del receptor de GnRH
- De síntesis de gonadotrofinas: genes de subunidades de gonadotrofina
- De acción de las gonadotrofinas: genes del receptor de gonadotrofinas.
En cuanto a los defectos de la migración de neuronas productoras de GnRH, el gen KAL se localiza en la región pseudoautosómica del cromosoma X (Xp22.3); codifica para Anosmin-1 y para la glicoproteína de matriz extracelular, encargadas del crecimiento y migración de neuronas olfatorias y productoras de GnRH; este gen origina el fenotipo del síndrome de Kallmann, un hipogonadismo hipogonadotrófico que se caracteriza por anosmia o hiposmia, fisura labiopalatina, sordera congénita, convulsiones cerebelosas, cuarto metacarpiano corto y agenesia renal, en 50% de los casos. El gen KAL se ubica en el cromosoma X y su expresión es fundamental los siguientes sitios (lo que explica las manifestaciones clínicas): cerebelo (sincinecia), núcleo óptico (anomalías visuales), mesénquima facial (defectos de la línea media facial) y meso-metanefro (agenesia renal). En cuanto a defectos en la síntesis y liberación de GnRH, no se han descrito alteraciones que provoquen alguna enfermedad en el ser humano; solamente se ha descrito mutaciones en el gen Gn RH con hipogonadismo hipogonadotrófico hereditario recesivo en animales de experimentación, específicamente en ratas.
La Fig. 1 es un esquema del gen del receptor de GnRH humano, en el cual se han descrito 14 mutaciones de tipo inactivante que pudieran condicionar casos de pubertad retrasada (1). Las mutaciones del gen del receptor de GnRH se han encontrado en 20% de los hipogonadismos hipogonadotróficos; la mayoría de estas mutaciones son heterocigotas y reducen, tanto la unión del GnRH con su receptor, como la activación de señales intracelulares, lo que produce fenotipos muy variables: la pérdida completa de la función se ve en la mutación Ala 1290 Asp / Ser 168 Arg, que da como manifestaciones fenotípicas micropene, criptorquidia, ausencia de desarrollo puberal y resistencia al tratamiento con GnRH pulsátil; otras mutaciones pueden dar compromiso leve de la función, con fenotipo de desarrollo puberal incompleto, gonadotrofinas basales detectables y modesta respuesta a GnRH pulsátil.
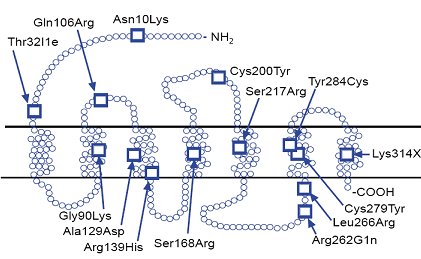 Tamaño completo
Tamaño completo Figura 1. Mutaciones en el gen del receptor del GnRH humano. Todas inactivantes (1)
En relación a mutaciones y polimorfimos en los genes de las subunidades alfa y beta de las gonadotrofinas, se sabe que la subunidad alfa es común para la hormona luteinizante (LH), la hormona folículo estimulante (FSH), la gonadotrofina coriónica (hCG) y la hormona estimulante del tiroides (TSH), de modo que una mutación en esta cadena tendría un resultado ominoso para el paciente. Las mutaciones de las subunidades beta para LH y FSH son las que se acompañan de pubertades retrasadas. Existe una tipificación en cuanto a los nucleótidos alterados y el intercambio de aminoácidos; se sabe que la mutación de la subunidad beta de LH puede provocar falta de inicio espontáneo de la pubertad y que cuando la mutación se ubica en el exón 2, se presenta un retraso en el inicio de la pubertad. Para la subunidad beta de FSH, algunas mutaciones en el exón 3 también pueden causar hipogonadismo masculino, con azoospermia e infertilidad y, en la mujer, amenorrea primaria e infertilidad (1).
La mutación del gen de la subunidad beta de LH se ha descrito solamente en un varón, que tenía genitales masculinos normales, pubertad retrasada, niveles de LH sérica elevada y FSH sérica normal, la cual aumentó con la edad; además, este paciente tenía espermatogénesis disminuida y ausencia de células de Leydig. Cuando se le realizó una prueba de estimulación con gonadotrofina coriónica se estimuló la síntesis de testosterona y la espematogénesis, pero no se logró conseguir fertilidad con esta metodología terapéutica.
Se ha descrito a cinco pacientes con mutaciones de la subunidad beta de FSH: tres mujeres adultas y dos varones, en quienes este gen se localizó en el brazo corto, fracción 13 del cromosoma 11 (11p13). En las mujeres, esta mutación produjo amenorrea primaria, infertilidad, ausencia de tejido mamario, pubertad retrasada con LH aumentada y FSH disminuida y presencia de glándulas suprarrenales normales. En los varones se presentó con pubertad normal, testículos pequeños, azoospermia, hipogonadismo secundario a déficit aislado de FSH, aumento de la LH sérica y disminución de la testosterona. Los casos señalados son situaciones infrecuentes, pero han permitido conocer cómo funciona esta cadena de genes en el desarrollo de la pubertad normal.
Las alteraciones de los receptores de gonadotrofinas a nivel gonadal se deben a mutaciones que se localizan en el cromosoma 2. Inicialmente se describieron mutaciones inactivantes en seis familias: en las mujeres había insuficiencia ovárica primaria, anovulación, amenorrea primaria o secundaria y presencia de folículos primordiales en el ovario; en los hombres las características sexuales secundarias eran normales, había grados variables de insuficiencia en la espermatogénesis y testículos pequeños, aunque ninguno de los pacientes era azoospérmico, la FSH sérica estaba moderadamente aumentada y la LH sérica estaba normal o moderadamente aumentada.
La causa más frecuente de pubertad retrasada, además de la constitucional, son las enfermedades crónicas, que pueden determinar que la pubertad se inicie en forma tardía. Existe una larga lista de este tipo de enfermedades (Tabla III).
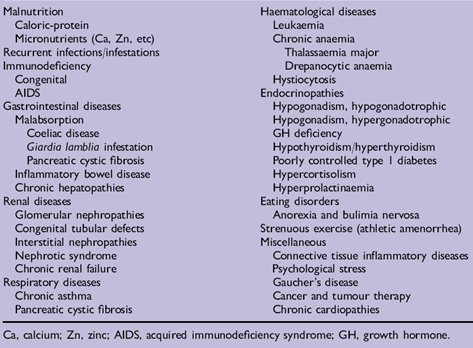 Tamaño completo
Tamaño completo Tabla III. Enfermedades crónicas causantes de pubertad retrasada
La malnutrición, probablemente por un mecanismo de conservación, afecta la producción de insulina, del factor de crecimiento insulinosímil (IGF-1) y de leptina y disminuye los niveles de hormonas tiroideas, específicamente T3 (2). Con más detalle, la disminución de la insulina aumenta la lipólisis y la proteolisis, probablemente porque el organismo busca una forma de compensación de fuente energética. La disminución de la síntesis de IGF-1 a nivel hepático se compensa con el aumento de la hormona de crecimiento, pero, como hay una disminución de la síntesis de receptores, esto determina un cuadro de resistencia a la hormona de crecimiento y retraso del crecimiento estatural. Por otro lado, si disminuye el tejido graso hay menos leptina, lo que ocasiona retraso de la pubertad. La T3 disminuida se traduce en disminución del metabolismo basal, de modo que este paciente esté en situación de conservación para un caso de estrés, en cuanto a nutrición (Fig. 2).
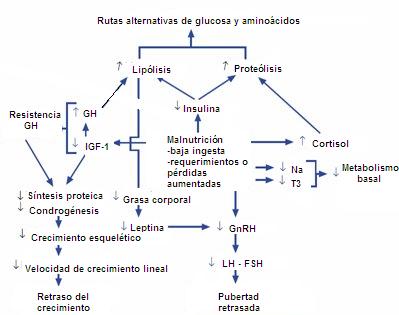 Tamaño completo
Tamaño completo Figura 2. Malnutrición y pubertad retrasada: fisiopatología
Estudio y tratamiento
En el estudio de una pubertad retrasada o detenida, el estudio mínimo incluye la determinación en sangre de: hormonas tiroideas, LH, FSH, testosterona o estradiol cuando corresponda, prolactina y exámenes generales para descartar una patología crónica, como hemograma-VHS, PCR, función renal, electrolitos plasmáticos y perfil hepático. En las mujeres se debe plantear la posibilidad de hacer un cariograma; en los hombres, éste se realiza según el fenotipo; en las niñas se debe hacer, además, una ecotomografía uterina y ovárica.Una vez que se han descartado las patologías mencionadas y se confirma que se trata de un retraso constitucional del crecimiento y la pubertad, se debe decidir si corresponde o no iniciar tratamiento. Por lo general se trata de un escolar varón, que está muy preocupado porque todos sus compañeros están creciendo rápidamente y él aún no ha dado el estirón puberal, ni ha cambiado la voz, ni tiene barba; con frecuencia se le han hecho varios tratamientos para el retraso puberal, con la idea de estimular un hipotálamo probablemente más lento y provocar el inicio de los cambios hormonales propios de la pubertad (3). En las niñas está situación es muy poco frecuente y los endocrinólogos infantiles la ven poco.
Algunas publicaciones señalan que algunos niños con pubertad retrasada tendrían compromiso de su estatura final, porque “no tienen tiempo suficiente” para alcanzar la estatura que les corresponde por su genética y se dice que esta pérdida, al final del crecimiento, sería de alrededor de 2 a 2,5 cms. Por lo tanto, cuando se habla de prescribir el uso de testosterona a un niño, sabiendo que es un esteroide sexual, que puede influir en un cierre anticipado de los cartílagos de crecimiento, es importante tener claro si es o no necesario su uso y si puede o no ser perjudicial. En un trabajo publicado en 2003 en Journal of Clinical Endocrinology, se tomó a 64 niños, separados en un grupo de 23 niños control y un grupo de 41 niños tratados con enantato de testosterona, en dosis de 125 mg intramuscular, durante tres meses. La talla y la edad de ambos grupos fue similar al inicio del tratamiento; las desviaciones estándar por debajo de la media también fueron semejantes y al término del crecimiento la talla de los niños fue muy similar, por lo que estos pacientes no se perjudicaron con un tratamiento que les permitió iniciar la pubertad en forma un poco más anticipada que lo que hubiera ocurrido de manera espontánea (4).
Cuando la pubertad retrasada se debe a una causa que impide la producción de esteroides sexuales, es necesario realizar la inducción de la pubertad, que en la actualidad ha mejorado bastante gracias a la disponibilidad de estrógenos de mejor calidad y la posibilidad de utilizar distintas vías de administración.
En las mujeres, la pubertad se puede inducir por distintos métodos: estrógenos conjugados de origen equino, etinilestradiol, 17 beta-estradiol y progestágenos, entre otros; todos los métodos son válidos según las condiciones de la paciente y siempre que se disponga de un tiempo de espera para cada una de las etapas, para remedar la pubertad normal. En la actualidad, el etinilestradiol es la forma menos onerosa de inducir la pubertad, de modo que es el método de elección en los hospitales del servicio público; también hay otras formas farmacéuticas, entre ellas cremas, geles y parches, que podrían lograr el efecto buscado. Básicamente, hay que tener en cuenta que la inducción se debe hacer imitando la pubertad normal fisiológica.
En los niños, en el Hospital Roberto del Río no hay experiencia en sustitución, sino con testosterona por vía parenteral. La masculinización con algunos preparados de testosterona administrados por vía oral es menos eficiente en cuanto a lograr una buena cantidad de vello masculino, buena masa muscular, etc., de modo que es más cómodo y menos costoso administrar testosterona por vía parenteral, como enantato.
Últimamente se está utilizando inhibidores de la aromatasa como tratamiento de la pubertad retrasada. En un estudio doble ciego, controlado con placebo, aleatorio, que incluyó a 23 varones con pubertad retrasada constitucional cuya edad media era 15 años, sin evidencias de estirón puberal a esa fecha, se dividió la muestra en tres grupos:
al primer grupo se le administró enantato de testosterona más letrozole; al segundo grupo se le administró enantato de testosterona y placebo y el tercer grupo fue el grupo control, es decir, no recibió tratamiento. En los grupos 2 y 3, que no recibieron inhibidor de la aromatasa, el 17 beta estradiol aumentó, mientras que en el grupo 1 se mantuvo, pero seis meses después de la suspensión del tratamiento, los niveles de estradiol se igualaron en los tres grupos. La testosterona subió en los tres grupos, ya que eran varones en etapa de crecimiento, pero en el grupo 1 el aumento fue mucho más exagerado a los cinco meses de tratamiento; al suspender el tratamiento, los niveles de testosterona también se igualaron en los tres grupos. A los cinco meses de tratamiento, en el grupo 1 aumentaron la LH y la FSH y aumentó la respuesta de LH a GnRH nativo, mientras que la respuesta de FSH a GnRH no varió. Los autores concluyeron que estos resultados demuestran que la inhibición de los estrógenos endógenos produce aumento de LH y FSH, a pesar de las altas concentraciones de andrógenos.
En el estudio aludido también se midió la inhibina B y se comprobó que hubo aumento en el grupo 1. Se midió además el eje somatotrófico, hormonas como la IGF-1 y la proteína transportadora IGFBF-3, las cuales aumentaron en el grupo 2, que sólo estaba recibiendo testosterona y se mantuvieron los niveles constantes en el grupo 1. Al suspender el tratamiento, en el grupo 1 se observó aumento de la velocidad de crecimiento, a los 18 meses de seguimiento; el grupo 2, que sólo recibió testosterona, creció solamente durante los cinco primeros meses de tratamiento. Además, se comprobó que el letrozole generó un retraso en la maduración ósea; por lo tanto, no hubo aumento de la talla en los pacientes que no fueron tratados con letrozole. Lo anterior sugiere que los andrógenos administrados en forma aislada no incrementan la talla adulta y confirma lo que se señaló sobre el enantato de testosterona en el crecimiento constitucional. Los resultados anteriores se correlacionaron con una mayor talla estimada para la adultez. De igual forma, se observó aumento de la talla estimada en todos los pacientes del grupo 1, en rango de 2,5 cms a 8,8 cms, excepto en un paciente. En cambio, en los pacientes de los grupos 2 y 3, que no recibieron el inhibidor de la aromatasa, no se modificó la talla estimada a los 18 meses de tratamiento (5).
Lo anterior hace suponer que la inhibición de los estrógenos, en los adolescentes en crecimiento, genera un aumento de la talla final; que no todos los pacientes con pubertad retrasada explotan su probable potencial genético de crecimiento; y que ése es el motivo por el cual la talla final de estos niños es algunos centímetros menor.
Referencias
- BPract and Res Clin End Met 2002; 16 (1):123-138.
- BPract and Res Clin End Met 2002; 16(1):73-90.
- Horm Res 2003; 60 (suppl 3):35-48.
- Clinical Endocrinology (2003) 58, 267-272.
- Wickman, Lancet 2001; 357:1743- 48.
