Cursos
← vista completaPublicado el 1 de junio de 2008 | http://doi.org/10.5867/medwave.2006.07.3420
MODY, otra forma de diabetes
MODY, another form of diabetes
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso Internacional de Diabetes del Niño y del Adolescente, organizado por la Asociación Latinoamericana de Diabetes entre los días 19 al 21 de abril de 2006. Directora: Dra. Gloria López.
Introducción
El término diabetes tipo MODY (Maturity Onset Diabetes in the Young) se acuñó, inicialmente, para un tipo de diabetes que se presenta en personas jóvenes, pero que no es dependiente de insulina. Hoy persiste esta denominación, pero se aplica a una diabetes congénita de herencia autosómica dominante, para la cual se han descrito por lo menos siete genes, de presentación variable en distintos grupos étnicos; es decir, un gen no se presenta en todas las poblaciones ni todas las variedades de diabetes monogénica que existen, se presentan en una misma población. La herencia dominante determina que se encuentre la enfermedad en más de 50% de los individuos afectados en tres generaciones, aunque muchas veces no se les reconoce como portadores de una diabetes monogénica. Algunos casos se califican como diabetes tipo 2 familiar y otros, como diabetes tipo 1 de alta incidencia familiar; pero cuando se hace un estudio específico de la familia, buscando mutaciones en los genes, se puede verificar que estos pacientes pertenecen a una variedad monogénica.
La diabetes monogénica, que puede ser tipo 1 o tipo 2, se debe a un defecto de intensidad variable en la síntesis de insulina, como ocurre en las variedades 3, 4 y 5 de la diabetes monogénica, o en la secreción de insulina, como sucede en la diabetes monogénica tipo 1 y 2. Está además la diabetes monogénica tipo 6, la que cursa con un estado de hiperinsulinemia que no se observa en las otras. Sea una diabetes tipo 1 o tipo 2, sobresale el hecho de que el número de células beta del páncreas no está alterado, es decir, no hay destrucción, autoinmune o no autoinmune de estas células; por lo mismo, los anticuerpos dirigidos contra el islote o contra la célula beta (ICA, IAA y GAD)son negativos en todas estas familias y habitualmente el cuadro se manifiesta antes de los 35 años de edad, aunque en algunos casos puede ser más tardío. Los pacientes tienen sensibilidad normal a la insulina y suelen tener peso normal; si hay sobrepeso, no es un factor determinante en la fisiopatología ni en la respuesta terapéutica. Los análisis de secretagogos primarios, como nutrientes, neurotransmisores y hormonas gastrointestinales, son normales, es decir, no hay alteraciones en la secreción de insulina frente a estas sustancias, como tampoco frente a los secretagogos potenciadores, como glucagón y arginina. Por último, a pesar de no ser insulinodependiente, en la casuística de México más de 50% de los individuos debutan con cetoacidosis diabética, por lo que se les puede confundir con diabetes tipo 1.
Mecanismos de secreción de insulina
La secreción de insulina (Fig.1) muchas veces está alterada en este tipo de diabetes. Para que la glucosa entre a la célula pancreática se necesita el transportador de glucosa tipo 2 (GLUT-2), cuya activación, además, libera glucosinasa, la que está unida a la membrana celular. Este paso es fundamental: para activarse, la glucosinasa se debe liberar del anclaje a la membrana celular, de tal manera que en el mismo momento en que la glucosa está entrando a la célula, es fosforilada (oxidada) por la glucosa-6P. Al mismo tiempo activa también una serie de cinasas, como la 2-6 glucosinasa (2-6 FFC) y la glutaraldehido deshidrogenasa (GDH). La oxidación de la glucosa, no por sí misma, sino como paso metabólico, causa dos alteraciones: por un lado, al final del proceso de oxidación de la glucosa aumenta la generación de ATP, con lo cual se cierran los canales de potasio y se despolariza la célula; y, al mismo tiempo, cuando la cantidad de ATP cumple con los requerimientos necesarios, se activa el ciclo de las NADH+H y se genera NAD reducido, que pasa a NADPH+H (fosfato). El aumento de estos dos intermediarios induce aumento en la cantidad de sulfhidrilos que están en el interior de la membrana celular, lo que prepara ese sitio para la secreción de insulina, mediante la activación de las cinasas A, B y C. Todo esto conduce a la formación de los túbulos y microfilamentos necesarios para formar el sitio específico de secreción de insulina.
Por otra parte, la oxidación de la glucosa causa una despolarización a la que colaboran el aumento de ATP y el cierre de los canales de potasio, por una parte, y el aumento de la actividad de la ATPasa, con la consiguiente disminución del ATP y del calcio en el interior de la célula, por otra. Esta despolarización se asocia con apertura de canales de sodio y calcio, con disminución de la salida celular de calcio; lo anterior favorece la unión de calcio con la calmodulina y se forma un complejo que disminuye la viscosidad y anula las cargas negativas de los gránulos de secreción, con esto se favorece la unión de éstos a la membrana, la que antes los rechazaba a causa de las cargas negativas. Los gránulos se unen al punto de la membrana que estuvo recibiendo el aumento de sulfidrilos, donde también se produjeron monofilamentos, y se unen a un sitio específico de secreción. Entonces, a partir de los mecanismos de oxidación de la glucosa, se selecciona el sitio y se favorecen los mecanismos para que se origine una secreción aguda de insulina.
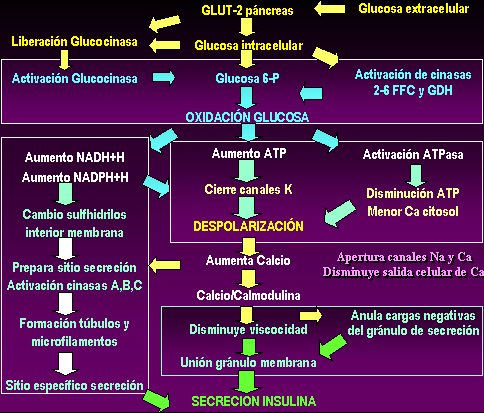 Tamaño completo
Tamaño completo Figura 1. Mecanismos de secreción de insulina
Tipos de diabetes MODY
En la diabetes MODY tipo 1, el patrón nuclear del hepatocito uno alfa (FNH 1-alfa) se encuentra alterado, lo que ocasiona dificultades con los mecanismos dependientes del transportador 2 de glucosa; este FNH 1-alfa regula la expresión de factores de transcripción de receptores de membrana que controlan la secreción hormonal que participa en el metabolismo de nutrientes; lo anterior altera los mecanismo iniciales de la oxidación y, además, impide la formación de túbulos y microfilamentos. Los dos mecanismos mencionados causan un defecto en la secreción de insulina, de tal manera que, frente a una carga de hidratos de carbono o de proteínas, se va a secretar una cantidad de insulina significativamente menor y va a aumentar la glicemia. El individuo es insulino-carente, ya que no tiene suficiente cantidad de insulina, por lo que semeja un cuadro de diabetes tipo 1, aunque no haya destrucción de la célula beta (Fig.2).
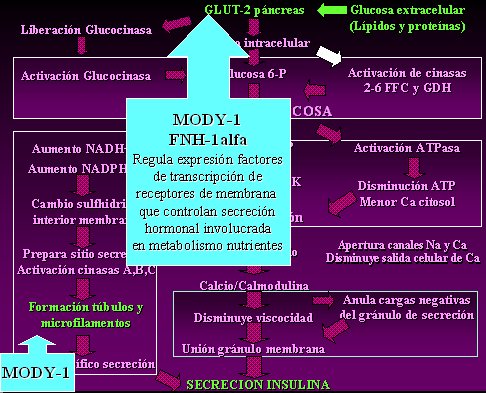 Tamaño completo
Tamaño completo Figura 2. Fisiopatología de la diabetes tipo MODY-1
En la diabetes monogénica tipo 2 hay una alteración específica en la activación de la glucosinasa, a tal grado que todos los demás procesos para la localización y la sensibilización de la insulina se encuentran alterados. Este fenómeno tiene menos efectos que la diabetes monogénica tipo 1 y, por lo tanto, muchos de estos diabéticos se comportan como si tuvieran diabetes mellitus tipo 2; o sea, la diabetes monogénica tipo 2 se puede comportar fácilmente como diabetes no dependiente de insulina (Fig. 3).
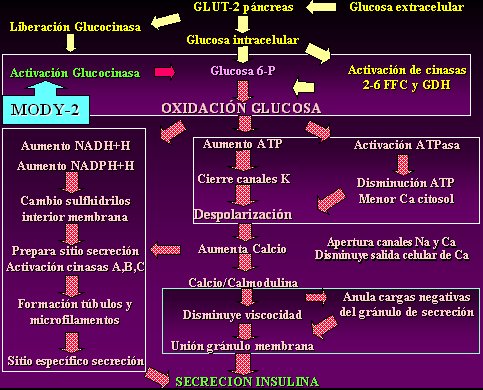 Tamaño completo
Tamaño completo Figura 3. Fisiopatología de la diabetes tipo MODY-2
En las diabetes monogénicas tipo 3 y 5, además de una alteración de la acción del transportador tipo 2 de glucosa en el páncreas, se encuentra alterado el último mecanismo, en donde los gránulos secretores van a anular cargas negativas y van a permitir la síntesis de insulina; pero además está alterada la síntesis de insulina, es decir, hay una insulinopenia real, además de la alteración de la secreción. Por lo tanto, las diabetes monogénicas 3 y 5 se comportan como diabetes totalmente dependientes de insulina, es decir, como diabetes mellitus tipo 1 (Fig. 4).
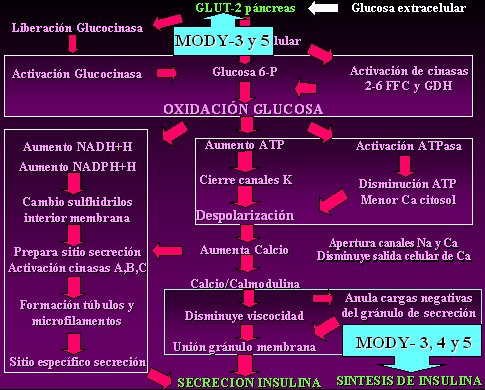 Tamaño completo
Tamaño completo Figura 4. Fisiopatología de la diabetes tipo MODY-3, 4 y 5
La diabetes monogénica tipo 1, en resumen, se caracteriza por la formación alterada de los microtúbulos, debido a un defecto en el brazo largo del cromosoma 20 (20q13.1) que codifica para el factor nuclear 4-alfa de los hepatocitos, el cual regula la expresión del factor 1 alfa (FNH 1-alfa), cuya alteración disminuye la expresión del GLUT-2 y la actividad de la aldosa beta reductasa, de la glucosa 3 fosfato deshidrogenasa (G3P-DH), particularmente en el páncreas, y de la piruvato cinasa, en el hígado. Los mecanismos de oxidación de la glucosa se encuentran alterados y, por lo tanto, la liberación de los gránulos de secreción de insulina también está disminuida. Cuando la alteración es grave, los pacientes se comportan como diabéticos tipo 1, dependientes de insulina (30%-50%); en cambio, cuando la alteración funcional es leve a moderada, se comportan como diabéticos tipo 2, lo que explica 0,25% de las diabetes tipo 2 en la población europea caucásica. Estos últimos evolucionan con frecuencia con daño microvascular rápido, pero no grave, o sea, como una diabetes aparentemente tipo 1, con un daño microvascular que evoluciona muy lentamente; en suma, como una diabetes no agresiva.
En la diabetes monogénica tipo 2, el defecto afecta la glucosinasa y se encuentra localizado en el brazo corto del cromosoma 7 (7p13-15). Es muy frecuente en Europa, donde representa alrededor de 12,5% de los casos de diabetes tipo 2; en cambio, en la población mexicana, tanto en la población mestiza con ancestros europeos como en la nativa, no se ha encontrado ni un solo caso de diabetes MODY tipo 2 entre las más de 400 familias de diabetes tipo MODY que se han analizado. La alteración de la glucosinasa altera también el receptor de glucosa y la fosforilación de la glucosa; pero, curiosamente, la secreción insuficiente de insulina se presenta sólo cuando la concentración de glucosa es inferior a 145 mg/dl. Cuando hay hiperglicemia moderada o grave, la secreción de insulina tiende a aumentar, por lo que los pacientes se controlan mucho mejor con medicamentos orales que mejoren la sensibilidad o potencien la secreción de insulina. Antes se creía que el efecto de los medicamentos ayudaba al paciente, pero lo cierto es que el mismo paciente es quien se ayuda, porque cuando la glucosa aumenta sobre 145 a 150mg/dl empieza a secretar mayor cantidad de insulina, lo que normaliza la concentración de glucosa y nuevamente surgen problemas de secreción, hasta que se elevan otra vez los niveles de glucosa.
Estos pacientes se comportan como una diabetes tipo 2 sin complicaciones micro ni macrovasculares y, cuando éstas llegan a existir, en caso de un control muy inadecuado del paciente, son de evolución muy lenta. No obstante, estudios europeos señalan que las mujeres heterocigotas hacen diabetes mellitus gestacional y que un porcentaje significativo de estos casos se deben a la variedad tipo 2 de diabetes monogénica. Cuando el recién nacido es homocigoto y presenta retraso del crecimiento intrauterino grave e hiperglicemia neonatal, la que tiende a ceder si el cuadro se detecta a tiempo y se hace un manejo adecuado. En ratones, cuando el defecto se localiza sólo en las células pancreáticas, se traduce en diabetes tipo dos; pero cuando otras células no pancreáticas sino también somáticas tienen el defecto, el cuadro es mucho más intenso y se comporta como una diabetes tipo 1 con todos sus componentes, lo que lleva a suponer que, en los seres humanos, la inmensa mayoría de los pacientes con diabetes tipo 2 son heterocigotos.
Las diabetes tipo 3 y tipo 5 se deben a distintos factores: en la primera está alterado el factor nuclear del hepatocito 1 alfa o FNH 1-a (12q24.2), en tanto que en la MODY-5 está alterado el 1 beta, o FNH 1-b (17q21.3), los que deben colaborar entre sí; por lo tanto, ambos tipos tienen un mecanismo etiopatogénico común, que es la alteración en la formación de homodímeros o de heterodímeros que regulan la transcripción de los genes del transportador 2 de glucosa (genes GLUT-2) y de esta gran familia de transcripción de la insulina. La consecuencia habitual es una disminución grave de la síntesis de insulina, la que se inicia en la etapa pospuberal y se manifiesta como una diabetes tipo 1 tardía que aparece después de la mitad de la vida, con un umbral renal bajo para la eliminación de glucosa, de manera que los pacientes tienen glucosuria con mucha facilidad y sin relación con el grado de hiperglicemia que hay en ese momento. Además, desarrollan rápidamente complicaciones renales y retinianas, a pesar de mantener un buen control, es decir, se comportan como una diabetes grave. Son pacientes que están relativamente bien controlados, con niveles de Hb glicosilada alrededor de 6 a 7, nunca sobre 7, pero a pesar de eso desarrollan nefropatía y retinopatía en los cinco o seis primeros años de evolución y estas complicaciones presentan una evolución rápida y de difícil manejo, a pesar de que se mejore en escala significativa el control metabólico.
En 6% de los casos, los pacientes con diabetes tipo 3 tienen buena sensibilidad a las sulfonilureas; por eso, cuando se utilizan estos fármacos, estos pacientes se comportan como una diabetes tipo 2, en cuanto a la mejoría del control de glucosa, pero un porcentaje muy alto de ellos no responden bien a las sulfonilureas y presentan microangiopatía de nervios y corazón, de evolución rápida. En algunas familias, las alteraciones coronarias se presentan después de los 20 ó 23 años de edad, y a veces no se sabe que el paciente tiene diabetes, sino que ella se diagnostica cuando se atiende el infarto. En la diabetes tipo 5, por alguna razón que aún no está clara, el número de nefronas es menor que lo normal y se forman quistes renales no congénitos, es decir, de tipo adquiridos, que condicionan hipertensión arterial sistémica y rápido desarrollo de insuficiencia renal crónica. En algunas familias se ha encontrado aplasia de vagina e hipoplasia grave de útero, lo que condiciona, obviamente, infertilidad, sin que se encuentre evidencia de lo que causa esta alteración. Lo importante es que en la diabetes tipo 5 es frecuente la alteración renal, aun en ausencia de microangiopatía retiniana, o sea, en esta población no hay una correlación, como se observa en la diabetes mellitus tipo 1 o autoinmune, entre retinopatía y nefropatía, sino que es mucho más frecuente encontrar nefropatía grave y rápidamente progresiva.
La diabetes tipo 4 se caracteriza por la alteración del factor promotor del gen de insulina, que está en el brazo largo del cromosoma 3 (IPF-1) (13q12.1). Los pacientes homocigotos tienen agenesia o hipoplasia grave del páncreas y se presentan como una diabetes neonatal, pero no en la forma clásica, en la que el paciente es hiperglicémico y luego tiende a controlarse, incluso sin tratamiento, sino como una diabetes agresiva que se presenta en menores de 3 meses de edad, en los que se comprueba ausencia total o hipoplasia muy grave del páncreas, sea in vivo o en la necropsia. En los parientes heterocigotos hay disminución moderada de la síntesis de insulina y ellos se comportan como diabetes tipo 2 de inicio postpuberal, con menor incidencia y prevalencia de microangiopatía, es decir, los heterocigotos tienen una diabetes tardía y los homocigotos, una diabetes tipo 1, muy agresiva y muy difícil de controlar.
La diabetes monogénica tipo 6 se presenta por alteración del factor de transcripción NEUROD-1/beta-2 (2q32), lo que se traduce en alteración de la síntesis de las proteínas que toman parte en el desarrollo del páncreas. Se comporta como una diabetes tipo 2 y es frecuente que los pacientes sean obesos e hiperinsulinémicos en ayunas, estado que se acentúa mucho tras la ingesta de alimentos y dificulta el diagnóstico diferencial entre la diabetes tipo 2 asociada a obesidad y la diabetes MODY tipo 6. En México hay sólo dos familias, a diferencia de otros países de América Latina, en los que la incidencia de obesidad ha aumentado de manera escalofriante. Hace doce años, 30% de los niños mexicanos que ingresaban a educación primaria a los 6 años de edad, tenían algún grado de desnutrición, por lo general leve o grado 1; hoy ya no se ve este problema, pero la transición epidemiológica ha llevado a que 26% de los niños que entran a primer grado sean obesos y casi todos ellos desarrollarán hiperinsulinemia y resistencia a la insulina. Un marcador clínico muy útil es la presencia de acantosis nigricans o pseudoacantosis nigricans: 99,3% de los obesos con este signo tiene resistencia a la insulina. Los portadores de diabetes MODY tipo 6 no desarrollan acantosis nigricans, aunque lleguen a ser muy obesos, y pueden tener hiperinsulinemia, pero sin resistencia a la insulina. Una de las familias era muy numerosa y se detectó porque varios de los niños consultaron por obesidad; tenían hiperinsulinemia, pero no acantosis nigricans y en su estudio se encontraron las alteraciones de la diabetes MODY tipo 6.
MODY en México
En un estudio realizado en México por un grupo multicéntrico se incluyó a pacientes del Instituto Nacional de Pediatría (INP) y de otros hospitales de tercer nivel. En los análisis genéticos se comprobó que 8% de los pacientes tenían anticuerpos anti-GAD 64 positivos en forma transitoria y que 100% tenían defectos en la secreción de insulina. Los genes fueron HNF-1a, HNF-4a, IPF-1, HNF-1b y NEUROD-1 y los tipos, MODY 1, 3 y 5; no se detectó ningún caso de MODY 2. Se trabajó con 60 familias y se observó que había mutaciones de estos factores, las que siempre afectaban la carga polar del gránulo de secreción o el tamaño de la cadena lateral, por lo que la secreción de insulina era extremadamente baja, a pesar de una síntesis normal, ya que no se anulaban las cargas negativas de los gránulos de secreción, de modo que no se podían fusionar a la membrana ni ser secretados. Ningún medicamento mejoraba estas alteraciones; sólo el poner al paciente en acidosis metabólica intensa mejoraba la secreción de insulina en forma transitoria, por 24 a 48 horas, pero después volvía a caer.
La edad de inicio del cuadro oscilaba entre los 12 y los 30 años de edad; 79% de estos pacientes necesitaban insulina para su control; predominaban los tipos 1, 3 y 5, con escasos casos de tipo 2; todos habían desarrollado cetosis, no cetoacidosis, en presencia de enfermedades intercurrentes; los diabéticos MODY tipo 3 también tenían microangiopatía en ojo, riñón o nervios periféricos, de presentación muy precoz, a los 5 años de evolución, y evolución rápida. Se observó obesidad en 70% de los individuos de estas 60 familias, lo que contrasta con lo que se describe en Europa, donde sólo 10% de las diabetes monogénicas se ven en pacientes obesos. La baja de peso mejoraba el control metabólico, pero no la dependencia de insulina, de tal manera que ésta no sería un factor de la génesis. Inicialmente, en 54% de los casos no se encontró mutaciones de los genes descritos en las diabetes monogénicas, pero después se encontró mutaciones nuevas en 37% de estos casos, dato que se publicó en su momento.
Genes MODY y diabetes tipo 2 de desarrollo temprano
Como en México está aumentando la incidencia de diabetes 2 en población joven, es importante saber si los genes MODY contribuyen al desarrollo temprano de diabetes tipo 2. Para responder a esta interrogante se estudió a 23 familias de diabéticos de inicio en edades muy tempranas, con 3 o más individuos afectados, además de 18 familias con diabetes tipo 2 y obesidad en 58% de los casos. Se analizaron los genes conocidos, es decir, HNF-1a, HNF-4a, IPF-1, HNF-1b y NEUROD-1 y se hizo un análisis de exones y regiones de unión exón-intrón. Lo que se observó con mayor frecuencia fue una heterocigosis en el HNF-1a exón 6 (P379H) que codifica para el dominio de transactivación de la proteína, y un polimorfismo HNF-1a (P394P, IVS6nt+27C), HNF-4a (A58A), IPF-1 (L54L) en un alto porcentaje de los pacientes que hicieron diabetes tipo 2. En la gran mayoría de ellos se presentó el cuadro en la etapa puberal inicial, con rango entre 12 y 38 años. Siempre tuvieron cetoacidosis diabética en el momento del diagnóstico: así debutaron y a los 12 años de evolución no tenían complicaciones microvasculares, ya que no es la misma población de diabetes MODY, sino una población de familias que iniciaban diabetes tipo 2 en forma precoz.
Para probar lo anterior, se transfectó el gen mutado HNF-1a (P379H) a células de cultivo (HeLa y RINm5f) y se demostró que así disminuía la actividad de transactivación del promotor de insulina en 25,9 y 35,7% respectivamente; además, se observó que no se afectó el nivel de expresión de la proteína. Se comprobó también que el polimorfismo IVS7nt G>A en el HNF-1 alfa es co-segregado con el fenotipo de diabetes mellitus en 80% de los pacientes y que cuando existe esa sucesión, hay algún grado de resistencia a insulina, tal como se había comunicado para G319S, detectado entre los oji-cree en 40% de los sujetos estudiados, lo que demuestra que la diabetes mellitus tipo 2 de presentación familiar temprana está asociada, aunque no se puede decir que haya una relación de causa-efecto, con la expresión de polimorfismos o de mutaciones del factor HNF-1 alfa.
En la actualidad, nuestros estudios están orientados a determinar cómo se comportan estas alteraciones genéticas. Se han observado familias en las que algunos de sus miembros afectados no tienen el polimorfismo o no tienen la mutación, pero hay miembros sanos que sí tienen el polimorfismo y lo segregan con las alteraciones en el HNF-1 alfa; así, no queda claro si estos factores son determinantes o son sólo acompañantes clínicos de alguna otra alteración. El hecho es que la diabetes monogénica, en el individuo menor de 18 años de edad, representa alrededor de 5% de los casos de diabetes insulinodependiente y 12% de los casos de diabetes no insulinodependiente; y en la población adulta, 12% de los individuos que se comportan como diabéticos tipo 2 son, en realidad, portadores de diabetes monogénica.
