Cursos
← vista completaPublicado el 1 de mayo de 2007 | http://doi.org/10.5867/medwave.2007.04.3449
Farmacocinética: absorción y distribución
Pharmacokinetics: absorption and distribution
Resumen
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso Actualización en Farmacología, organizado por Medwave Capacitación Limitada y la Unidad de Capacitación del Hospital Padre Hurtado entre los días 9 y 30 de junio de 2003. Director del Curso: Dr. Juan Diego Maya.
Absorción y vías de administración
La velocidad de absorción se puede modificar por factores propios del fármaco o por factores propios del sitio de absorción.
Entre los primeros están la liposolubilidad y la constante de disociación, es decir, si se ioniza o no. También está la concentración o dosis, que está dada por la cantidad que existe en el tiempo cero de aplicación en el sitio de la administración y que determina el gradiente que va a empujar la droga a través de la membrana. Si se administra una cantidad muy pequeña, es probable que ese gradiente no se establezca o que dure muy poco. Finalmente, es importante la forma farmacéutica, que puede limitar la velocidad de la absorción. Por ejemplo, existen muchas formas farmacéuticas que se pueden administrar por vía oral: solución, jarabe, presentaciones en gel y sólidas, que incluyen cápsulas, tabletas, comprimidos, pastillas y otras; cada una de ellas tiene una velocidad de desintegración distinta. Es obvio que la solución no se desintegra, ya está disuelta, por lo tanto la velocidad de absorción de una solución es mucho mayor que la de un comprimido, que primero se debe desintegrar, lo que en general ocurre en el estómago, para que la forma activa se disuelva en la fase acuosa intestinal y se absorba. La vía sublingual es variable; por ejemplo, cuando se aplica nifedipino se punciona la perla y se aplica, lo que equivale a administrar una forma líquida, mientras que el Isordil (isosorbide) sublingual es una forma sólida.
Entre los factores propios del sitio de absorción está la superficie de absorción; el intestino es un ejemplo muy claro, ya que su gran superficie permite la rápida absorción de medicamentos, al igual que los pulmones. La piel tiene el inconveniente de que es una barrera y que las sustancias deben ser extremadamente liposolubles para absorberse; los esteroides fluorados son muy liposolubles y por eso se absorben por vía dérmica, mientras que la hidrocortisona tiene muy mala absorción. La irrigación del tejido es otro factor importante, que explica porqué la Aspirina, por ejemplo, no se absorbe en el estómago, porque este órgano tiene poca irrigación, a diferencia del intestino; mientras mayor es la velocidad del flujo sanguíneo en el intestino, mayor es la velocidad de absorción, lo que ocurre en el estado postprandial. Muchos medicamentos se podrían absorber más rápido sobre las comidas que antes de las comidas. Algunos se tienen que absorber en ayunas, pero es por otras razones.
La velocidad de vaciamiento gástrico y la velocidad de tránsito intestinal también afectan la velocidad de absorción; si el fármaco pasa con rapidez por el intestino, no alcanza a entrar en contacto con la mucosa y sale intacto del tubo digestivo. La presencia de enzimas metabolizantes es otro de los factores influyentes: si en la mucosa intestinal existen estas enzimas, el fármaco no alcanzará la sangre, ya que será inactivado por éstas. Por ejemplo, la adrenalina por vía oral no se absorbe debido a la presencia de la monoaminoxidasa (MAO) intestinal, que la degrada rápidamente; si no hubiese MAO intestinal se podría administrar por vía oral. Lo mismo pasa con la insulina, es una proteína que es degradada por las enzimas pancreáticas. En cambio, el neosintrón, que es un anticoagulante, se absorbe por vía oral sin problemas, tanto así que se llama anticoagulante oral, no tiene el problema de que las enzimas pancreáticas o intestinales lo digieran.
Muchos medicamentos pueden ser transportados por un grupo de transportadores inespecíficos, como la glicoproteína P, que transporta todas las moléculas que encuentra y se ubica en múltiples sitios. En el intestino, estos transportadores se ubican desde la luz intestinal hacia la sangre y se encargan de ingresar sustancias; en la barrera hematoencefálica miran hacia la cara cerebral y extraen sustancias desde el cerebro hacia la sangre, ya que aquí no entra nada; en el riñón están mirando hacia la sangre, para extraer sustancias hacia la orina, es decir, la función depende de la ubicación. En las células neoplásicas también existen y pueden sacan los fármacos antineoplásicos desde la célula hacia la sangre, lo que constituye un mecanismo de resistencia a estos fármacos.
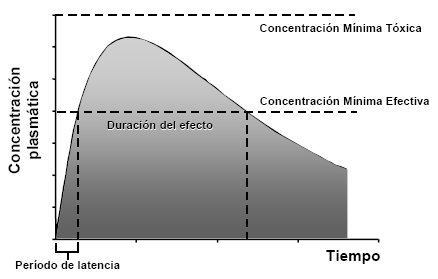
Cuando se administra un fármaco por vía oral se establece una concentración en función del tiempo (la farmacocinética siempre es en función del tiempo). La Figura 1 ilustra la evolución de las concentraciones plasmáticas en función del tiempo, lo que explica varios procesos en forma simultánea: el de absorción, de distribución y, por último, el de eliminación. A tiempo cero, cuando el paciente toma la tableta o el comprimido, no hay droga en el sistema, por lo que la concentración plasmática es igual a cero; a medida que se van estableciendo los elementos absortivos van aumentando las concentraciones plasmáticas. En la curva, esta velocidad de aumento es muy rápida al principio, ya que el incremento en la concentración plasmática ocurre en un período muy corto debido al enorme gradiente que impulsa la droga hacia la sangre, hasta que se llega a una meseta, que está dada por el equilibrio entre la cantidad de fármaco que está ingresando al organismo y la que está siendo eliminada, por lo que la concentración plasmática se estabiliza. Luego, en el intestino se va acabando el fármaco, por lo tanto la absorción disminuye, de modo que comienza a predominar el fenómeno fisiológico de la eliminación y las concentraciones plasmáticas comienzan a descender. La eliminación no es tan rápida como la absorción.
Además de la descripción de la curva, existen en ésta varios elementos descriptivos, como el período de latencia, que es el tiempo que demora el fármaco en alcanzar la concentración terapéutica mínima y, por lo tanto, el tiempo que demora en ejercer su efecto. La concentración plasmática sigue subiendo por sobre este nivel, pero después de un tiempo desciende y vuelve a quedar por debajo; si el síntoma que se quiso tratar desapareció no es necesario repetir el medicamento, pero si persiste se debe volver a administrar, aunque con condiciones muy claras. Lo importante es que la dosis del fármaco no provoque una velocidad de absorción tan grande que haga que las concentraciones plasmáticas sobrepasen la línea punteada, ya que esto produciría concentraciones tóxicas. En resumen, en la curva se observa la latencia, la duración del efecto, que es el período en que se alcanzan las concentraciones terapéuticas mínimas y un techo, que es la concentración mínima tóxica. Esto, en dosis única; con dosis sucesivas existen muchos elementos que complican la situación.
 Tamaño completo
Tamaño completo Figura 1. Curva de nivel plasmático en función del tiempo cuando se administra una dosis única por vía oral
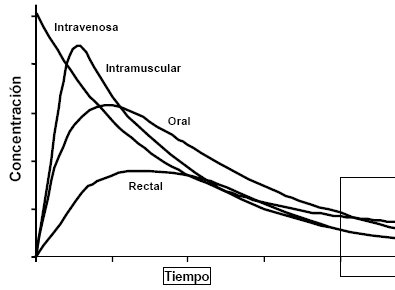
La ruta afecta la velocidad de administración; la que proporciona mayor velocidad de absorción es la endovenosa, porque permite obtener las concentraciones plasmáticas en forma instantánea, a menos que el fármaco se administre en bolo lento, pero eso es por otras razones. La vía intramuscular no proporciona concentraciones plasmáticas instantáneas debido al retardo del proceso de la absorción, pero como el sitio de administración está muy cercano a los vasos sanguíneos la velocidad de absorción es bastante alta y depende tanto del volumen como del sitio de la aplicación; no es lo mismo una inyección en el deltoides que en el muslo, porque éste tiene más irrigación. Si se hace masaje aumenta la irrigación y se acelera la absorción, las que también depende del tamaño muscular, que permite mayor o menor volumen. La vía oral es lenta, pero es cómoda, barata y tiene otra ventaja adicional: si se administra el fármaco en exceso se puede retirar, lo que no se puede hacer con las vías venosa, intramuscular o subcutánea.
Por vía rectal la curva es más irregular que como se representa en la Figura 2, porque la absorción rectal es errática y puede dar concentraciones muy altas o subterapéuticas en el mismo paciente y con la misma presentación; sin embargo, en ocasiones es la única vía disponible, como ocurre con un niño que convulsiona y en el que no se logra obtener una vía venosa, en cuyo caso se puede administra diazepam por vía rectal, con buena absorción, aunque de todos modos errática. Esto se debe a que la ampolla rectal tiene doble irrigación: en la mitad inferior el drenaje venoso va a dar a la circulación sistémica, es decir, a la vena cava y de ahí al resto del organismo; en cambio, la porción superior de la ampolla rectal y sigmoides drena a la vena porta, por lo tanto el fármaco debe pasar primero por el hígado antes de llegar a la circulación sistémica. Las proporciones de fármaco que alcanzan una u otra irrigación nunca son constantes, por lo que la absorción es errática y por esta causa no es la mejor vía, pero a veces es la única disponible. Funciona mejor con fármacos que tienen un índice terapéutico muy amplio.
 Tamaño completo
Tamaño completo Figura 2. Relación entre nivel plasmático y vía de administración
Biodisponibilidad
El concepto de biodisponibilidad es fundamental para entender los procesos que ocurren tras la administración oral de un fármaco. La biodisponibilidad es el porcentaje de fármaco administrado que alcanza la circulación sistémica; si se administra a un paciente un fármaco por vía oral, parte de ese fármaco se va a perder en forma insoluble, otra parte va a ser metabolizada en el intestino, otra parte va a ser metabolizada en el hígado y sólo una fracción va a alcanzar la sangre: ésa es la fracción biodisponible, es decir, la fracción capaz de ejercer el efecto. Cuando se ingiere un comprimido de propanolol de 80 mg para controlar la hipertensión arterial, solamente 25% de esa dosis alcanza la circulación plasmática y el resto se metaboliza en el hígado, pero este efecto metabólico hepático ya está calculado, de modo que ese 25% que alcanza la circulación sistémica es suficiente para provocar el efecto terapéutico. Por lo tanto, la biodisponibilidad depende, en primer lugar, del grado de absorción.
La biodisponibilidad depende también de la eliminación de primer paso, la que depende del metabolismo intestinal o hepático. Eso explica que la nitroglicerina se deba administrar por vía sublingual, ya que en este caso el efecto de primer paso es cercano a 90% y el 10% restante no es suficiente. Las presentaciones comerciales de Isordil sublingual son mucho menores que las de Isordil mantención, que toman los pacientes cada ocho horas, porque el que se va a tomar por vía oral tiene que tener una dosis muy alta para provocar el efecto, por el efecto de primer paso; la vía sublingual permite evitar ese efecto y llegar más rápido al sistema donde se desea que actúe el fármaco. En cuanto a la vía inhalada, en algunos casos de reanimación hay que instilar adrenalina por el tubo endotraqueal, porque llega de inmediato al corazón y no se va a metabolizar, aunque la adrenalina es un mal ejemplo, porque no se administra por vía oral, pero existen muchos otros fármacos, como el salbutamol, que si se administra una dosis excesiva igual llegan a la circulación sistémica, aun cuando el salbutamol oral se metaboliza en el hígado. Las dos estrategias que se pueden utilizar para sobrepasar el efecto de primer paso son: utilizar una vía diferente o elevar la dosis.
Distribución
Una vez que el fármaco ingresa al torrente sanguíneo, queda en el mejor vehículo para que lo distribuya a lo largo y ancho del organismo. Hasta el momento no hay fármacos inteligentes, es decir, la Aspirina se distribuye por igual en todo el organismo, aunque la persona tenga el dolor en la cabeza o en el hombro.
Los factores que afectan la distribución son los mismos que ya se han descrito: liposolubilidad; grado de ionización; irrigación sanguínea, es decir, si un órgano tiene una alta irrigación sanguínea el fármaco se va a distribuir en él, de preferencia. Otros factores que afectan la distribución son, la unión a las proteínas plasmáticas y la afinidad por los tejidos. En el plasma y los tejidos los fármacos van acompañados de las proteínas en mayor o menor proporción y se establece una competencia entre las proteínas del plasma y las proteínas tisulares; la distribución del fármaco va a depender de quién gane en esa competencia: si las proteínas tisulares atraen con mayor facilidad o avidez al fármaco, éste no va a permanecer en la sangre, sino que va a pasar a los tejidos; si, por el contrario, la afinidad del fármaco por las proteínas de la sangre es mayor, se quedará allí.
El fármaco que está unido a las proteínas plasmáticas y tisulares es farmacológicamente inerte, a menos que las proteínas tisulares sean los receptores; pero si está atrapado por otra proteína indiferente, no funciona. Un ejemplo es la digoxina, que tiene una gran afinidad por las proteínas tisulares y se acumula en el músculo esquelético, que es mucho más grande que el corazón; sólo una pequeña porción de la digoxina va a llegar a las proteínas receptoras que existen en el corazón y determinan su efecto terapéutico, el resto se distribuye en el músculo esquelético, en forma homogénea, ya que la digoxina tampoco es inteligente, y no ejerce función alguna. La afinidad por los tejidos determina el fenómeno de acumulación y permite explicar el parámetro de volumen aparente de distribución.
La distribución no siempre es uniforme debido a que hay barreras, de las cuales la principal es la barrera hematoencefálica. Se habla de la barrera placentaria, pero en realidad ésta no existe; si así fuera, las mujeres embarazadas podría tomar cualquier medicamento, en cambio sólo pueden tomar penicilina y paracetamol, porque todo lo demás pasa al niño. Lo que sí es cierto es que la placenta tiene actividad metabólica, por lo que puede metabolizar en cierta medida algunos fármacos, pero no lo suficiente como para constituir una verdadera barrera, como la barrera hematoencefálica.
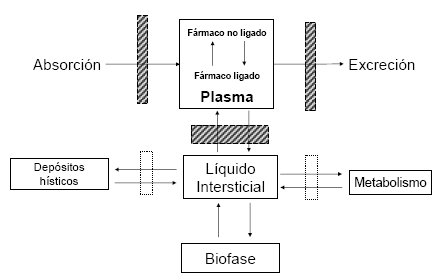
La distribución tiene distintos tiempos y sigue un modelo multicopartimental (Fig. 3). La absorción va a llevar el fármaco al compartimiento general, central, que es la sangre y eso incluye también a los órganos de alta perfusión: corazón, pulmón, cerebro e hígado, donde se va a establecer un equilibrio dinámico entre lo que hay en la sangre y lo que hay en esos órganos; pero también hay un equilibrio dinámico entre lo que hay unido a las proteínas y lo que está libre, porque el fármaco no está unido 100% a las proteínas, algo tiene que estar libre en la sangre y pasar a los tejidos; lo que se mueve es el fármaco libre, no el unido a las proteínas. El fármaco que sale a los tejidos ingresa al tejido intersticial, donde establece interacciones con las proteínas correspondientes y algunas de esas proteínas pueden ser las encargadas de mediar su mecanismo de acción; también puede pasar a depósitos hísticos, por ejemplo, el músculo es depósito hístico de la digoxina.
 Tamaño completo
Tamaño completo Figura 3. Modelo de distribución bicompartimental
Cuando se administra tiopental a un paciente para inducir anestesia, el fármaco dura en el organismo 96 horas en promedio, aunque su efecto dura minutos. ¿Cómo se explica que el tiopental, a pesar de durar 96 horas, no deje al paciente anestesiado durante 96 horas? Esto se explica porque, una vez que se inyecta el tiopental, éste va a los órganos de alta perfusión, como el cerebro y, como es muy liposoluble, no tiene problemas con la barrera hematoencefálica: llega, entra y actúa, pero también se va rápido, porque la irrigación sanguínea lo lava con rapidez y lo lleva hacia el tejido adiposo, donde se queda guardado, sin funcionar; luego sale con lentitud hacia la sangre, continúa hacia el riñón y, finalmente, se elimina, por eso dura tanto tiempo. En otras palabras, existen fenómenos de distribución, uno rápido y uno lento; el fenómeno de distribución rápida ocurre en un compartimiento que cumple con la característica de rápida irrigación; el lento tiene lenta irrigación, por lo tanto las concentraciones plasmáticas que va a mantener el tiopental desde ahí son muy bajas.
Lo anterior ilustra el hecho de que la distribución inicial del fármaco depende del flujo sanguíneo de cada tejido y de la capacidad de acumularse en ese tejido; pero, independiente de si la irrigación es alta o baja, la salida del fármaco depende de la permeabilidad y de la afinidad por el tejido. Si el fármaco es muy liposoluble, tendrá afinidad por el tejido lipídico. Sin embargo, la afinidad es reversible, es decir, el fármaco que pasó a los tejidos y se encontró con su proteína no va a quedar pegado para siempre, porque daría un efecto a perpetuidad; la afinidad es reversible y depende de la concentración plasmática, la cual depende, a su vez, de la velocidad con que se está absorbiendo y de la velocidad con que se está eliminando el fármaco. Si se está eliminando rápido, lo que está en los tejidos se va a la sangre para poder recuperar el depósito sanguíneo, con la consecuencia final de que va a ser excretado.
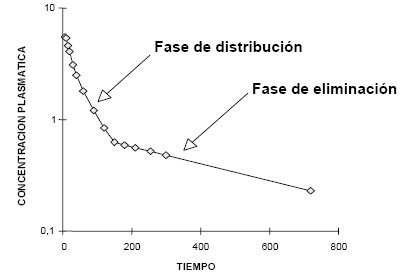
En la Figura 4 se ilustra lo que ocurre cuando se aplica un fármaco por vía intravenosa, en bolo: se adquieren concentraciones plasmáticas altas, o máximas, en forma instantánea, debido a que el fármaco se diluye instantáneamente en todo el organismo; desde ese momento la concentración plasmática baja rápidamente, ya que el fármaco se está distribuyendo, saliendo de la sangre a los tejidos; como la concentración plasmática inicial es mucho más alta, debe salir hacia los tejidos, esté unido o no a las proteínas.
 Tamaño completo
Tamaño completo Figura 4. Modelo de distribución bicompartimental. Gráfico semilogarítmico de las concentraciones plasmáticas en relación con el tiempo
El fármaco se distribuye primero en los tejidos de más alto flujo; después, cuando se alcanza el equilibrio entre la concentración plasmática y la concentración tisular, porque cesa la administración del fármaco, comienza a predominar el fenómeno de eliminación, que es un fenómeno mucho más lento, porque hay muy poca droga en el plasma, de modo que la velocidad con que se elimina el fármaco es lenta. Esta fase no sólo incluye la eliminación, sino también la distribución a los tejidos poco perfundidos; por ejemplo, el tiopental pasa del cerebro al tejido adiposo, lo que también ocurre con lentitud, porque se establece un equilibrio entre tejido adiposo y plasma, lo que amortigua la caída de las concentraciones plasmáticas, ya que lo que cae por eliminación es reemplazado por lo que sale del tejido adiposo.
