Cursos
← vista completaPublicado el 1 de mayo de 2005 | http://doi.org/10.5867/medwave.2005.04.3548
Mecanismos de acción, reacciones adversas y nuevos antimicóticos
Mechanisms of action, adverse reactions and new antifungal agents
Resumen
Este texto completo es la transcripción editada y revisada del Curso de Actualización en Micología Médica, Infecciones Fúngicas Invasoras y Nosocomiales, organizado por el Instituto de Ciencias Biomédicas (ICBM) de la Universidad de Chile entre los días 29 de noviembre al 1 de diciembre de 2004.
Director del Curso: Dr. Víctor Silva, MSc.,PhD.
Coordinadora del Curso: Dra. Lily Contreras.
Edición científica: Dr. Víctor Silva.
Introducción
A continuación se tratarán los mecanismos de acción desde el punto de vista microbiológico. Al conocer cómo funcionan los hongos y hacia qué estructuras van dirigidas los antimicóticos, es posible deducir las reacciones adversas que éstos generan.
Sitio de acción de los antifúngicos
Las levaduras son eucariontes; por lo tanto, se diferencian mucho de las bacterias en cuanto a su estructura, la que presenta varios sitios donde pueden actuar los antimicóticos (véase figura 1).
En la mayoría de los casos, el fármaco antimicótico actúa en la membrana citoplasmática del hongo, específicamente en la síntesis de ergosterol; esto ocurre, por ejemplo, con la familia de los polienos, a la que pertenecen la anfotericina B y la nistatina, y con la familia de los azoles, que son los fármacos más utilizados en clínica. La familia de las alilaminas, entre las cuales destaca la terbinafina, también bloquea la síntesis de ergosterol.
Hace unos años, se utilizaba mucho la griseofulvina, que actúa sobre la división nuclear e inhibe la mitosis, inhibiendo los microtúbulos, razón por la cual su toxicidad es importante en las células del torrente sanguíneo; por eso, su uso debía ser controlado estrictamente con hemograma. Por otra parte está la fluorocitosina, un análogo de nucleósido que es capaz de inhibir la síntesis de ADN y ARN.
Durante años se estuvo buscando un fármaco selectivo para las células fúngicas, que no actuara sobre las células humanas, de modo de disminuir las reacciones adversas; hasta que se descubrieron las equinocandinas, que inhiben la síntesis de la pared celular del hongo, de la cual carecen las células mamíferas, siendo drogas mucho más selectivas.
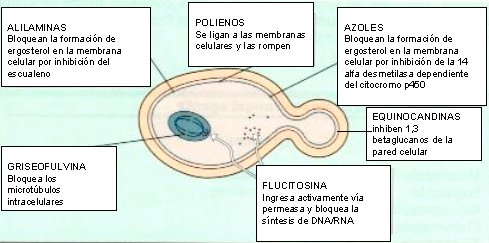 Tamaño completo
Tamaño completo Figura 1. Sitio de acción de los antifúngicos.
El ergosterol es un componente lipídico de la membrana sobre el cual actúa la mayoría de los fármacos antimicóticos. Es el esterol que predomina en las células fúngicas y, entre sus funciones, da fluidez e integridad a la membrana, permite la función apropiada de muchas enzimas unidas a ella y, al favorecer la función de la quitina sintetasa, permite el crecimiento y la división celular. Las levaduras y los hongos filamentosos presentan, generalmente, yemaciones o células hijas, por lo que es preciso que la membrana sea bastante dinámica. La síntesis del ergosterol, cuyo precursor es el escualeno, está compuesta por una serie de etapas, como se ve en la figura 2.
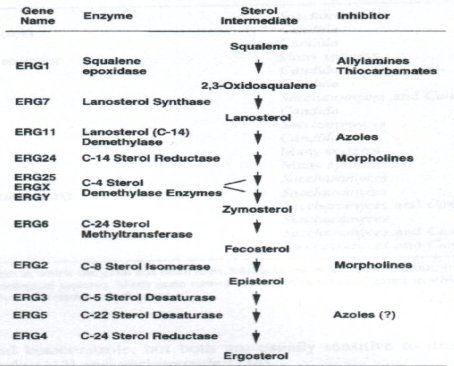 Tamaño completo
Tamaño completo Figura 2. Vía de la biosíntesis de ergosterol, a partir de escualeno.
Los antimicóticos como los azoles y la amorolfina, que se utiliza mucho en micosis de uña como tratamiento tópico, actúan en ciertas etapas de la síntesis del ergosterol; inhibiendo específicamente algunas enzimas, por ejemplo, las alilaminas bloquean la escualeno epoxidasa y los azoles bloquean la 14 alfa lanosterol desmetilasa. Estas enzimas son codificadas por una familia de genes que pueden mutar y generar una resistencia secundaria a los antimicóticos.
La pared celular es otro sitio de acción importante. Es una estructura muy compleja, compuesta en 90% por polisacáridos y en 10% a 20% por lípidos y glicoproteínas. Del punto de vista morfológico, determina las distintas formas que tienen los hongos; además, les permite interactuar con el ambiente y protege a éste contra la lisis osmótica, es un sitio de unión para enzimas y tiene propiedades antigénicas, que son aprovechadas para realizar diagnósticos.
A continuación se revisarán las principales características de los antimicóticos.
Polienos
Los polienos más importantes son la nistatina, que se usa en forma tópica, y la anfotericina B, que se administra por vía intravenosa. Son de estructura lipídica, lo que dificulta el suministro; incluso, cuando se realizan pruebas in vitro hay que disolverlos con solventes específicos; por eso también es difícil utilizarlas en el tratamiento de pacientes.
Esta familia se une a los componentes de la membrana plasmática e interactúa con el ergosterol, pero sin inhibir su síntesis. Al unirse al ergosterol, se forma un poro de gran tamaño por el que se pierden iones, azúcares y otros compuestos, hasta que la célula finalmente revienta. Por eso, y además porque la unión es irreversible, la anfotericina es un fármaco fungicida.
En cuanto a la especificidad de los polienos, las bacterias no tienen ergosterol, sino otros componentes, por lo que son insensibles a estos compuestos, sin embargo, este fármaco, podría interactuar con la membrana de las células eucariontes, razón por la cual la anfotericina B es bastante tóxica. La estructura química de la anfotericina B es similar a la del colesterol, por eso tiene afinidad por la membrana, se adhiere a ella y ocasiona su rotura.
Los distintos antifúngicos que componen la familia de los polienos tienen distinta afinidad por lípidos; por ejemplo, la anfotericina B tiene más afinidad por el ergosterol que por el colesterol. Antes había algunos antifúngicos, como las filipinas, que presentaban mayor afinidad por el colesterol. Cuanto más afinidad tenga un fármaco por el colesterol, tanto más tóxico es para el ser humano.
La anfotericina B es bastante tóxica, genera reacciones febriles, gastrointestinales y azotemia, por nefrotoxicidad; por lo tanto, siempre que se suministre este fármaco se debe monitorizar los niveles de potasio, la función renal y las reacciones febriles, y utilizar antipiréticos cuando sea necesario; además, puede ocasionar anemia normocítica normocrómica. Para disminuir los efectos adversos se han creado formulaciones liposomales o emulsiones lipídicas en las cuales la anfotericina, que es insoluble en agua, va dentro de la emulsión. Estos productos solamente mejoran la tolerancia, porque la potencia es muy similar a la del fármaco puro.
En cuanto a su espectro, la mayoría de los hongos oportunistas se inhiben con concentraciones relativamente bajas de anfotericina B. Antes se planteaba que esto ocurría bajo 1 ug/ml, pero es muy difícil establecer puntos de corte tan bajos al medir la susceptibilidad a este agente. En algunas especies se ha planteado que sobre 0,5 ug/ml podría haber algún grado de resistencia. Por eso, este valor de CIM en la actualidad se utiliza más como un punto de referencia.
Algunos agentes, como Trichosporon, Scedosporium y algunos hongos dematiáceos (feohifomicosis) son intrínsecamente resistentes a la anfotericina B, debido a sus características estructurales. Por eso es importante realizar siempre un buen diagnóstico micológico, porque si un paciente tiene un cuadro clínico característico y se le aísla un hongo negro o una feohifomicosis, el tratamiento con anfotericina B no va a ser eficaz, ya que muchas de estas especies son intrínsecamente resistentes.
La resistencia primaria a anfotericina B no es muy frecuente; se ha empezado a ver resistencia secundaria, pero muy poco, porque es tan eficaz como fungicida que el hongo no alcanza a realizar cambios que le permitan adaptarse a este fármaco. Sin embargo, se ha visto que algunas especies son capaces de sustituir su componente de ergosterol por precursores más cercanos al lanosterol y defenderse de esta manera.
También se ha visto inducción de cepas resistentes por mutagénesis, química o por luz ultravioleta; cuando se administran tratamientos quimioterápicos. La célula fúngica es capaz de recibir este mismo tóxico o noxa, y, de manera secundaria, empieza a defenderse de los citotóxicos generando cambios en su contenido de ergosterol. El hecho de que cambie el componente de ergosterol de su membrana no siempre significa que la célula va a ser resistente, sino por el contrario, muchas veces la célula resultante es más vulnerable a otros cambios ambientales.
En general, las cepas aisladas resistentes a polienos tienen menor cantidad de ergosterol. Este fenómeno se ha visto en algunas cepas de Candida albicans, Saccharomyces cerevisiae, Aspergillus spp. y en Neurospora crassa. Sin embargo, en algunos mutantes aumentan la cantidad de ergosterol y son resistentes, como se ha visto con cepas de Candida albicans y C. neoformans; o sea, no existe una norma que determine si el cambio en el contenido de ergosterol va a generar resistencia.
Como conclusión, la modificación o ausencia de esterol en la membrana no es la única explicación bioquímica de la resistencia; por ejemplo, en S. cerevisiae se ha identificado 6 genes participantes en resistencia a polienos, pero el estudio de esta resistencia no está muy desarrollado, porque es poco frecuente.
5 fluorocitosina
La 5 fluorocitosina (5FC) es una pirimidina fluorinada, cuya molécula tiene un átomo de flúor que actúa inhibiendo la síntesis de ADN y ARN en la célula fúngica. Es un fármaco bastante específico, que no actúa en la síntesis de nucleótidos de las células humanas. Este antimicótico no está disponible en Chile, pero en otros países se utilizó mucho, en combinación con anfotericina B y con algunos fármacos azólicos.
La 5 fluorocitosina puede entrar en la membrana fúngica gracias a la enzima citosina permeasa, que no está presente en la célula de mamíferos, por eso es específica. Al entrar en la célula, esta molécula sufre modificaciones y va hacia la síntesis de ARN, pero origina una fluorouridina aberrante, o sea, genera un ARN mutante, que va a afectar la síntesis proteica, función esencial en las células. Por otra parte, puede inhibir la enzima que permite el paso de uridina a timidina, o timidilato sintetasa, porque el ADN está constituido por timidina y no por uracilo, lo que también afecta la síntesis de ADN (véase Figura 3).
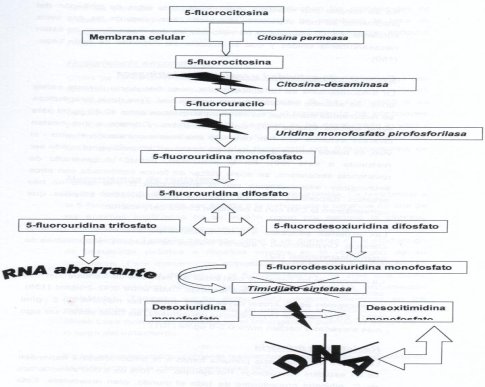 Tamaño completo
Tamaño completo Figura 3. Mecanismo de acción de la 5 fluorocitosina.
Aunque este fármaco es muy tóxico para la célula micótica, el hongo se defiende mutando determinadas enzimas que participan en su acción, como la citosina permeasa, cuya mutación impide la entrada del antimicótico, de modo que éste no tendrá efecto; además puede alterar enzimas que forman parte de la vía de modificaciones que sufre el fármaco de la célula, como la urodina monofosfato pirofosforilasa. Este fármaco rápidamente genera resistencia secundaria; es decir, una vez que la célula fúngica ha estado en contacto con él comienzan a generarse todas estas mutaciones y se presenta la resistencia.
Lo anterior tiene muy buena correlación in vitro/in vivo , por lo que, cuando aparece una concentración inhibitoria mínima alta, igual a 32 ug/mL o mayor, el hongo es bastante resistente a la terapia. Los niveles plasmáticos recomendados son de 40 a 60 ug/mL, para evitar toxicidad.
La mayoría de las especies de Candida albicans son sensibles a 5FC (CMI = 0,12-2 ug/mL), con excepción de C. krusei (CMIs> 8 ug/mL), la que además presenta resistencia a fármacos azólicos. Una gran ventaja es que se puede absorber vía oral y en general los niveles plasmáticos se alcanzan con relativa facilidad. Este antimicótico es activo contra Candida albicans, Cryptococcus y algunos hongos dematiáceos, pero tiene el inconveniente de su rápida generación de resistencia secundaria. Otra ventaja que tiene la 5 fluorocitosina es que presenta sinergismo con polienos y drogas azólicas como anfotericina B, ketoconazol y fluconazol, lo que permitiría bajar la dosis de la anfotericina B y disminuiría la toxicidad para el paciente.
Azoles
Estos fármacos cambiaron la historia de la medicina y se considera que los antifúngicos sistémicos derivados del imidazol y del triazol constituyen el avance más importante de los últimos años, en el tratamiento de las micosis sistémicas oportunistas. La aparición del fluconazol, después de la anfotericina, permitió lograr una disminución de la toxicidad de los tratamientos antimicóticos. Los imidazoles son bastante tóxicos, pero los triazoles sirven para tratar algunas micosis sistémicas que antes eran intratables, con muy buena tolerancia por parte de los pacientes.
El grupo de los azoles está compuesto por dos familias: los imidazoles y los triazoles, que comparten mecanismos de acción y resistencia. Entre de los imidazoles están clotrimazol, miconazol y ketoconazol; este último era uno de los fármacos más utilizados en la onicomicosis, pero su toxicidad hepática es muy importante.
Los triazoles se toleran mejor; entre de ellos destacan fluconazol e itraconazol, que son los triazoles de primera generación; recientemente se desarrollaron los de segunda generación, entre ellos voriconazol, ravuconazol y posaconazol, cuyo espectro de acción ha mejorado frente a otros hongos. El que se ha estudiado más ha sido el voriconazol, con muy buenos resultados en cuanto a muerte celular.
En comparación con los más antiguos, estos fármacos son activos contra más especies de Candida albicans y, además, son activos contra Aspergillus, a diferencia de los azoles de primera generación, con excepción del itraconazol, que antes se utilizaba en la aspergilosis invasora. Voriconazol ha mejorado el pronóstico de las aspergilosis invasoras y su perfil de seguridad es muy similar al de los triazoles, con escasas reacciones adversas. La aparición de estos nuevos azoles se debe al trabajo científico que se ha realizado para disminuir al máximo la toxicidad de los fármacos y mejorar la potencia de drogas, con el mismo mecanismo de acción.
Los azoles se pueden administrar por vía oral y parenteral, se distribuyen relativamente bien y se disuelven con facilidad. En cuanto a sus mecanismos de acción, inhiben la biosíntesis de ergosterol en el paso de desmetilación del lanosterol, en el carbono 14; por lo tanto, su blanco es la lanosterol-14-alfa-desmetilasa, que es una de las especies del citocromo p450, localizada en el retículo endoplasmático. La disminución del ergosterol más la acumulación de esteroles metilados causa una alteración de la membrana celular, la que se vuelve más permeable y vulnerable a daños.
De ahí deriva la toxicidad de estos fármacos, porque algunos de los azoles son menos selectivos y suelen actuar sobre enzimas hepáticas y causar hepatitis fulminantes, porque el mecanismo descrito compromete funciones que dependen de la membrana, en la que puede haber otras enzimas. Así, se altera el transporte de nutrientes y la síntesis de quitina, lo que puede inhibir el crecimiento.
Los azoles no siempre son fungicidas; depende de su tipo. Los imidazoles tienden a ser fungicidas en concentraciones altas, pero en general se acepta que son fungistáticos y que su efecto es limitado, aunque la situación ha cambiado con los nuevos fármacos, porque son más potentes sobre las células. El hecho de ser fungistático se relaciona directamente con la capacidad de desarrollar resistencia secundaria, porque siempre quedan poblaciones que no mueren con el fármaco y comienzan a generar mecanismos para defenderse de este fármaco.
La toxicidad presenta una diferencia importante entre los dos grupos de azoles, debido a su selectividad. En consecuencia, los imidazoles son más tóxicos. Por ejemplo, el ketoconazol inhibe la síntesis de testosterona en concentraciones 100 veces más bajas que el fluconazol; a su vez, hay menos actividad intrínseca de los triazoles ante Candida albicans y se necesita CMIs más altas; por eso, muchos de estos fármacos, como el clotrimazol, se utilizan en formulaciones tópicas y pueden tener un efecto fungicida en el sitio de infección.
Los efectos adversos son náuseas, anomalías endocrinológicas (irregularidad menstrual, ginecomastia en hombres) y alteraciones de las pruebas hepáticas, que es uno de los efectos más frecuentes. El ketoconazol es el más tóxico. Cuando se administran estos fármacos se deben monitorizar las pruebas hepáticas y ajustar la dosis en pacientes que sufren daño hepático o que están usando otras drogas que podrían interferir. Actualmente se recomienda el uso de azoles menos tóxicos (triazoles).
El espectro de actividad es amplio, tanto en los triazoles como en los imidazoles, los que actúan sobre levaduras, dermatofitos, Aspergillus y otros hongos filamentosos; el fluconazol es la excepción, pues carece de actividad sobre algunas levaduras (C. krusei) y sobre Aspergillus, Sporotrix, Rhizopus y otros hongos filamentosos.
Por eso, siempre se debe identificar la especie de Candida albicans y cuando aparece una infección importante, invasora, en la cual sólo se ha determinado el género, no se puede iniciar el tratamiento con un azol, lo ideal es partir siempre con anfotericina B y después, según la especie, se puede cambiar a azol. En caso de carecer de test de susceptibilidad, la decisión se toma sobre la base de los datos locales o publicados, aunque en realidad la mayoría de las Candida albicans son susceptibles.
Otros hongos que muestran resistencia intrínseca, o primaria, frente a los azoles son el Scedosporium prolificans y algunas especies de Candida albicans, que son hongos ambientales, pero que, por el mismo motivo, están en contacto estrecho con tóxicos y estructuralmente presentan resistencia intrínseca. Candida glabrata y Candida guillermondii son levaduras haploides; es decir, tienen un alelo para un gen, a diferencia de Candida albicans, que tiene dos, de manera que si muta uno de los alelos el otro puede suplir la función del mutado.
La resistencia secundaria no se pesquisó hasta la década de 1980, cuando pacientes con candidiasis mucocutánea crónica (por alteración de su respuesta inmunitaria innata) necesitaron tratamientos periódicos con algunas drogas azólicas, en especial ketoconazol, y presentaron fracasos terapéuticos y recaídas con los usos profilácticos prolongados de antimicóticos, como en el caso de la candidiasis orofaríngea en el SIDA.
En 1986 se describió el primer caso de resistencia secundaria de Candida albicans a ketoconazol, que era la droga que más se utilizaba, en ese momento, en un paciente con SIDA.
En cuanto a los mecanismos de resistencia, el gen ERG11 codifica para la enzima blanco de los azoles; por tanto, uno de los mecanismos de resistencia del hongo es mutar este gen. No se trata forzosamente de mutaciones por estar en contacto con el tóxico; se sabe que ciertas poblaciones de Candida albicans normalmente van sufriendo cambios, pero cuando surge una presión de selección, estos cambios se aceleran y surgen algunas poblaciones que se defienden mejor contra el agente.
Otro mecanismo de defensa consiste en modificar la diana mediante alteraciones genéticas que aumentan la expresión del gen o lo amplifican, o efectúan conversión o recombinación mitótica, que son mecanismos más complejos, pero lo importante es que pueden aumentar la producción del lanosterol y competir con el antimicótico.
Además, en todas las células eucariontes y bacterias existen transportadores o bombas de flujo, que son proteínas relativamente conservadas en todas las especies y, frente a determinados tóxicos, son capaces de expulsarlos al exterior de las células (Ej: existe una bomba de expulsión de azoles). Estas estructuras se encontraron al analizar células de pacientes que no respondían a quimioterapia y se determinó que pertenecen al grupo de transportadores ABCT, codificados por los genes CDR. El gen MDR1 codifica para una proteína de la familia de los facilitadores principales, un tipo de bomba que aparece en la membrana celular.
Por ejemplo, en un paciente con SIDA se analizaron las cepas de distintos episodios de infección y se descubrió que, en una cepa que tenía una determinada concentración inhibitoria mínima, aumentaba este parámetro frente a un segundo episodio, aunque correspondía a la misma población clonal. Entonces se analizaron cuáles eran las mutaciones que realmente eran relevantes en este caso y se encontraron algunas mutaciones que sólo estaban presentes en las células que presentaban la concentración inhibitoria mínima más alta.
Para que las mutaciones causen resistencia, tienen que ocurrir en sitios activos de la enzima, es decir, en los sitios de unión con el antimicótico; constituyen las llamadas mutaciones hot (calientes), que ocurren donde se produce el efecto in vitro. Por otra parte, para determinar si un gen se expresaba más, se midió el ARN mensajero del gen de las bombas de flujo o del ERG11, usando como control ARN ribosomal, que siempre se está expresando en una cantidad constante; se vio que, en algunas cepas cuya concentración inhibitoria mínima estaba aumentada, se sintetizaba más ARN mensajero para estos genes (véase figura 4).
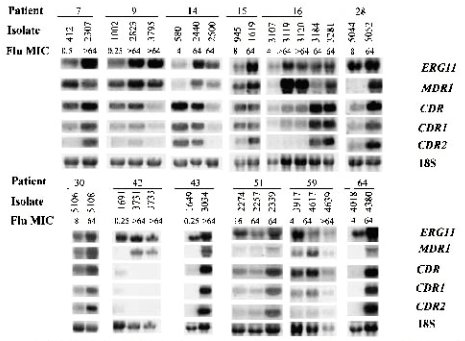 Tamaño completo
Tamaño completo Figura 4. Northern blots de RNA total de aislados de Candida albicans analizados con sobdas radiomarcadas específicas para ERG11, MDR1, CDR, CDR1 y CDR2 (Antimicrob Agents Chemoter, 2001; 45: 2676-2684).
En el modelo de resistencia secundaria que se ve en la figura 5 se observa que el fármaco azólico entra en la célula y un número reducido de bombas lo exportan hacia afuera, pero el fármaco logra llegar a la enzima blanco, la 14 alfa lanesterol desmetilasa. Cuando la administración del tóxico es constante, el hongo sintetiza más ARN mensajero, se sobreexpresan las bombas y comienza a expulsar la droga hacia afuera, pero además puede mutar el ERG11; en distintos estudios se ha tratado de ver cuál es el mecanismo determinante, y no se ha logrado observar predominancia de alguno; en general, todos están funcionando.
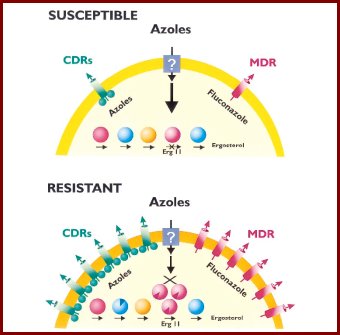 Tamaño completo
Tamaño completo Figura 5. Modelo de resistencia secundaria a azoles (Clin Microbiol Rev, 1998; 11: 382-402).
La importancia clínica de la resistencia a azoles radica en lo siguiente:
- En las candidiasis vaginales, están apareciendo especies menos sensibles, C. tropicalis y C. glabrata, de las cuales la segunda es una de las más importantes. Algunos estudios han demostrado que las candidiasis recurrentes se asocian con cepas que tienen estos mecanismos de resistencia.
- El uso profiláctico de fluconazol en pacientes neutropénicos y pacientes con pancreatitis ha producido un aumento de infecciones por C. krusei que no responden a tratamiento. Hubo una tendencia a usar drogas azólicas de manera profiláctica en la pancreatitis, por el riesgo de infección, pero esta práctica condujo a que se seleccionaran cepas resistentes.
- Las candidiasis superficiales en inmunosuprimidos son crónicas y por eso dichos pacientes reciben tratamiento prolongado, lo que puede generar cepas resistentes de Candida albicans.
La aparición de resistencia secundaria ha obligado a cambiar las indicaciones de profilaxis; cada vez se utiliza menos el tratamiento profiláctico, el que se reserva sólo para pacientes con riesgo muy elevado y con dosis más altas, para evitar que sobrevivan poblaciones de levaduras que más adelante generen resistencia secundaria y fracaso terapéutico.
Griseofulvina
El mecanismo de acción de la griseofulvina consiste en inhibir la mitosis de los hongos con la producción de células multinucleadas. Esta droga interrumpe el huso mitótico e interactúa con los microtúbulos polimerizados. Es fungistática in vitro para varias especies de dermatofitos. No tiene efecto sobre bacterias ni sobre otros hongos, levaduras, Actinomyces ni Nocardia, pero en general, en una época, dio muy buenos resultados en el tratamiento de las dermatomicosis. Tiene el inconveniente de que se debe administrar por períodos muy prolongados, de seis meses a un año. Además, como es poco selectiva, hay que controlar su uso con hemograma. Por estos motivos, hoy está en desuso.
Alilaminas: terbinafina
La terbinafina actúa por inhibición de la enzima escualeno epoxidasa en la membrana celular micótica, por lo que también altera la síntesis de esteroles, conduce a déficit de esterol y acumulación de escualeno causando la muerte celular micótica. Por eso es tan eficaz en la onicomicosis; tres meses de tratamiento bastan para erradicar el hongo, lo que no sucedía antes con otros antimicóticos.
Está demostrado que las onicomicosis generan efectos psicológicos importantes en los pacientes, quienes no se atreven a usar sandalias; es una enfermedad muy molesta. En bajas concentraciones, la terbinafina es fungicida contra dermatofitos, mohos y algunos hongos dimórficos; en las levaduras, según la especie, puede ser fungicida o fungistática. De hecho, se ha utilizado también en otras terapias.
Además, la terbinafina es relativamente selectiva y no presenta tantos efectos adversos como las otros antimicóticos, porque la enzima que inhibe no está relacionada con el sistema citocromo p450. No influye en el metabolismo hormonal y, en general, no se ve afectada por otros fármacos de metabolismo hepático. Se puede administrar por vía oral o tópica, pero de todas maneras es preciso ajustar la dosis en pacientes con patología hepática o renal. Se tolera bien y permite acortar el tratamiento de la onicomicosis.
Equinocandinas
En general, la mayoría de los fármacos antimicrobianos provienen del ambiente y son sintetizados por hongos y bacterias como los Streptomyces. Muchos hongos están en contacto con estos agentes constantemente y por eso algunos son intrínsecamente resistentes. En España se encontró una equinocandina en el ambiente: la caspofungina, que cambió la historia de la aspergilosis invasora en cuanto a la mortalidad, ya que la disminuyó de 100% a 50%.
Este fármaco actúa en la pared celular del hongo, inhibe la síntesis de glucanos, componentes que no están en la célula eucarionte humana, por lo que es relativamente selectivo. Las principales indicaciones de administración son las candidiasis y las aspergilosis invasoras; no se describen muchas reacciones adversas. También están en el mercado la micafungina y la anidulafungina.
Nuevos antifúngicos
En la actualidad hay fármacos nuevo que actúan sobre los factores de elongación en la síntesis del ARN e inhiben la síntesis proteica, como la sordarina; otros inhiben la síntesis de quitina, como la nikkomicina y algunos péptidos antimicrobianos. A medida que se avanza en el conocimiento de la estructura del hongo se puede apuntar a nuevos blancos en forma selectiva.
En resumen, el propósito de esta exposición fue incorporar el concepto del sitio de acción de los antimicóticos, porque es distinto al de otros antimicrobianos.
