Notas metodológicas
← vista completaPublicado el 8 de abril de 2020 | http://doi.org/10.5867/medwave.2020.02.7869
Conceptos generales en bioestadística y epidemiología clínica: estudios experimentales con diseño de ensayo clínico aleatorizado
General concepts in biostatistics and clinical epidemiology: Experimental studies with randomized clinical trial design
Abstract
In experimental studies, researchers apply an intervention to a group of study participants and analyze the effects over a future or “prospective” timeline. The prospective nature of these types of studies allows for the determination of causal relationships, but the interventions they are based on require rigorous bioethical evaluation, approval from an ethics committee, and registration of the study protocol prior to implementation. Experimental research includes clinical and preclinical testing of a novel intervention or therapy at different phases of development. The main objective of clinical trials is to evaluate an intervention’s efficacy and safety. Conventional clinical trials are blinded, randomized, and controlled, meaning that participants are randomly assigned to either the study intervention group or a comparator (a “control” group exposed to a placebo intervention or another non-placebo or “active” intervention—or not exposed to any intervention) to reduce selection and confounding biases, and researchers are also unaware of the type of intervention being applied. Intention-to-treat analysis (inclusion of all originally randomized subjects) should be done to avoid the effects of attrition (dropout) and crossover (variance in the exposure or treatment over time). A quasi-experimental design and external controls may also be used. Metrics used to measure the magnitude of effects include relative risk, absolute and relative risk reductions, and numbers needed to treat and harm. Confounding factors are controlled by randomization. Other types of bias to consider are selection, performance, detection, and reporting. This review is the fifth of a methodological series on general concepts in biostatistics and clinical epidemiology developed by the Chair of Scientific Research Methodology at the School of Medicine, University of Valparaíso, Chile. It describes general theoretical concepts related to randomized clinical trials and other experimental studies in humans, including fundamental elements, historical development, bioethical issues, structure, design, association measures, biases, and reporting guidelines. Factors that should be considered in the execution and evaluation of a clinical trial are also covered.
Main messages
- Randomized clinical trials evaluate the efficacy and safety of therapeutic interventions, allowing causality to be established.
- There are several types of randomized clinical trials.
- Randomized clinical trials effectively control confounding bias; however other specific biases may occur.
- International guidelines regulate ethical conduct for executing these studies.
Introduction
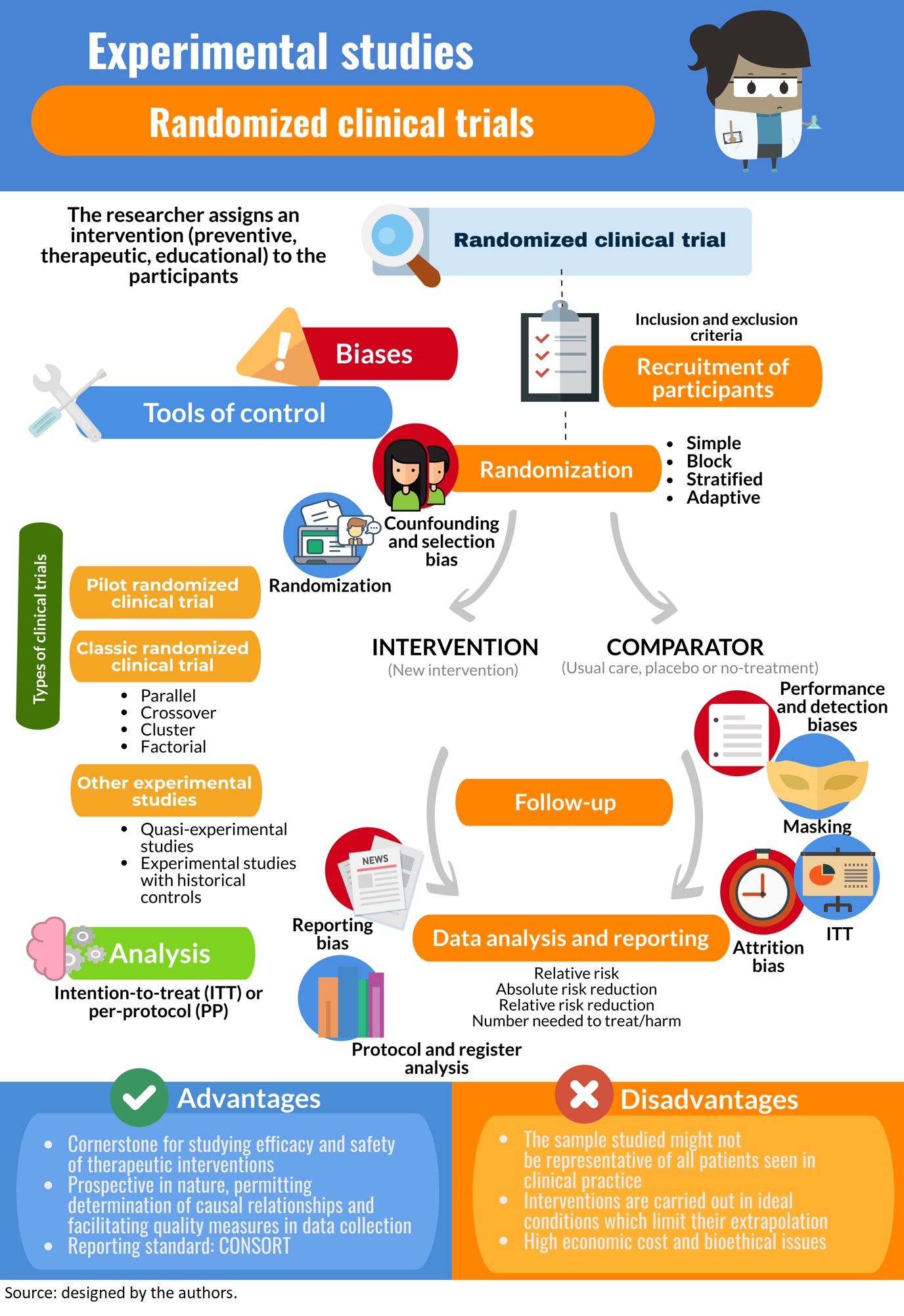
Experimental studies are those in which the researcher applies an intervention to the participants, as defined in a previous article of this methodological series[1]. This is the framework for all types of clinical trials that analyze preventive, therapeutic, educational, among other interventions, and that might be carried out on individuals or population groups[2],[3]. Some authors consider case series without a control group as a starting point for studies on therapeutic interventions, since they contribute to the development of new surgical techniques and the development of interventions in very rare conditions where a clinical trial would be difficult to undertake[4]. However, randomized clinical trials are the methodological design-of-choice for assessing efficacy (the true biological effect of an intervention) and effectiveness (the effect of an intervention in everyday clinical practice)[5]. This design provides the greatest ability to control biases[6].
In the 1920s, Ronald Fisher conceptualized randomization after he applied a random assignment of treatments or varieties to field plots in agricultural experiments. Later, in a study published in 1948[7], the Medical Research Council in the United Kingdom adapted randomization to clinical epidemiology by conducting a randomized clinical trial to evaluate the effect of streptomycin among pneumonia carriers, generally recognized as the first randomized clinical trial[8]. However, as early as 1907, the physician William Fletcher had published the results of a randomized clinical trial conducted to analyze the origin of beriberi in patients admitted to a psychiatric asylum in Kuala Lumpur[9], where conditions could be better controlled. He assigned patients to eat white or brown rice, according to theory at the time that associated beriberi with consumption of white rice.
Over the last 70 years, clinical trials have been refined and have become the fundamental methodology of regulatory drug agencies for authorizing the marketing of pharmaceutical products[10]. Research into the harmful potential of some drugs is relevant in light of public health issues: sudden death in patients anaesthetized with chloroform[11],[12], long-bone aplasia in newborns from mothers treated with thalidomide for pregnancy related nausea and vomiting[13], and, more recently, adverse effects of acetylsalicylic acid[2],[14]. Several national entities thus focus the greater part of health research funding budgets toward the execution of clinical trials[10]. Example 1 presents a randomized clinical trial.
This article is the fifth in a methodological series of six narrative reviews on general topics in biostatistics and clinical epidemiology, which explore published articles available in major databases and specialized reference texts. The series is aimed at training undergraduate and postgraduate students and is carried out by the Scientific Research Methodology Chair at the School of Medicine of the University of Valparaíso, Chile. The aim of this manuscript is to address the main theoretical and practical concepts of experimental studies in humans, primarily in the form of randomized clinical trials.
Preliminary concepts: control, randomization, and blinding
Clinical trials correspond to prospective experimental designs (a follow-up is made) that afford the ability to establish causal relationships given the trials corroborate that the cause (intervention) precedes the effect (outcome). A "controlled" trial implies results in the intervention group are compared to results in a "control" or comparator group, yielding a statistical estimate of the effect size. If a control group were not included, it would not be certain that the outcome is attributable to the intervention[5]. It is thus necessary to include at least two groups of patients and/or healthy volunteers that are randomly assigned to an experimental treatment or a control, hence the qualifier "randomized" clinical trials. In the process of randomization, neither the researcher nor the experimental subject is involved. Randomization is a key phenomenon in this type of design, as it is the principal means for controlling key biases associated with human research. In fact, randomization has been considered by some authors as the "most revolutionary and profound discovery of modern medicine," since multiple great discoveries have achieved clinical use through its application: from the onset of penicillin to gene therapy[8].
Controlled clinical trials include those that are open or "unblinded," where the participant and the researcher are both aware of the assigned intervention (for example, in randomized clinical trials that evaluate surgical interventions). This was shown in Example 1, where in a first "open" phase, all participants were aware of using an antidepressant for 16 weeks. In contrast, "blinding" implies subjects do not know the treatment arm they were assigned. Traditionally, the terms "single blind," "double blind" and "triple blind" referred to the blinding of participants, the blinding of researchers, and the blinding of the evaluators of the principal outcomes, respectively. However, the terms caused confusion as to exactly who was blinded, and for the sake of clarity, it is considered best practice that all groups blinded are specifically reported[16]. "Masking," on the other hand, refers to the same process, but more specifically to hiding by means of a "disguise;" for example, if the intervention is a drug that is administered in a tablet, the tablets of the intervention group (active ingredient) and comparison group (placebo) will have essentially the same characteristics. Some studies have progressed further in maintaining blinding and employ "active placebos" that mimic the experience of taking the intervention. For instance, if a drug generates dry mouth, participants may realize they are receiving the active ingredient. An active placebo may also generate the effect of a dry mouth, but not produce the effect related to the active ingredient[17]. In conclusion, both blinding and masking are related to the same principle[3],[18],[19],[20],[21]. Example 2 presents an open-label randomized clinical trial.
In reporting of the results of a randomized clinical trial, we often see a so-called "Table 1," where relevant biosociodemographic characteristics are reported, such as sex, age, socioeconomic level, comorbidities, relevant concomitant therapies, among others. It has a descriptive but also analytical value, since it allows comparison of the baseline characteristics between the groups.
Classification of clinical trials
Although clinical trials are typically associated with drug development, this design allows the evaluation of any type of intervention. The Food and Drug Administration (FDA), the regulatory agency of the United States, classifies clinical trials in phases according to their stage in the developmental pipeline for a pharmaceutical product, as presented below. These phases are often misrepresented in the literature, and the terms have also been used in trials examining nonpharmacological interventions[2],[5],[10],[16],[23].
- Pre-clinical studies. These studies detect safety problems of the drug product, such as carcinogenicity and teratogenicity. They involve processes such as chemical synthesis, biological testing, and toxicological studies. They are performed within experimental models, principally in animals.
- Phase I trials. These are pharmacological studies without therapeutic objectives that principally evaluate toxicity, pharmacokinetic and pharmacodynamic parameters, tolerance, responses at different doses, and maximum safe dose. They are typically performed in healthy volunteers, or possibly patients with advanced disease with no other possibility of treatment, where it might not be appropriate to treat volunteers. Therefore, these represent the first tests on humans. They are usually open-label and uncontrolled, and less than 100 participants are involved.
- Phase II trials. These correspond to the first clinical exploration of the treatment, where the most appropriate posology for phase III studies must be defined. They provide preliminary information on efficacy and clinical safety and are carried out in patients with the disease under study. There may or may not be a control group, but if it is employed, random allocation is applied. Phase II trials usually include between 100 and 300 participants. They may involve an early phase (II a), typically consisting of pilot studies to evaluate the safety and activity profile of the new intervention (primarily bioavailability), and a later phase (II b), which typically aims to guarantee aspects such as safety and the possible superiority of the intervention over an existing one. Phase II studies can act as a screen for drugs with real potential to be evaluated in phase III.
- Phase III trials. Their aim is to demonstrate the effect of an intervention under conditions similar to those that can be expected when the drug is widely available (therapeutic confirmation studies). They are usually conducted in multiple centers, typically involving more than 300 participants (sometimes thousands). They are known as "pivotal" or "confirmatory," as a sample size associated with a statistical significance is calculated previously, and the trial proceeds toward demonstrating the hypothesis with a pre-established level of statistical power. While toxic effects must have been investigated in phases I and II, frequent side effects will be determined in phase III, indicating the types of patients particularly susceptible to them. Phase III trials are required for registration and to authorize the marketing of a novel pharmaceutical product.
- Phase IV trials. These evaluate the drug in wide-spread clinical use, hence further variety in the types of patients treated is likely to occur. They provide additional information on risks, adverse events, benefits, new uses, long-term effects (pharmacovigilance), drug interactions, among others. They are carried out after approval and marketing of the pharmaceutical product (post-marketing). Phase IV studies may involve an observational study design (for example, case series, case-control studies, cohort studies), and thus are subject to the biases inherent in these designs. However, they provide important information about the application of the drug or intervention in the "real world."
Regardless of whether the clinical trials study pharmacological or non-pharmacological interventions, they can be classified as unicentric, defined as performed by a single research group at a single center, or multicentric, when a common research protocol is executed by more than one research group at more than one center. The latter allows for the study of a larger number of participants in less time, with more reliable and representative conclusions on the population; however, their planning, coordination, monitoring, management and data analysis is more complex[16],[23].
Finally, it is common to find the term "pilot study" for certain clinical trials in the published literature (Example 3)[24]. These correspond to preliminary trials whose objective is to carry out a survey in order to execute a subsequent clinical trial of greater relevance. Pilot studies provide insight into the accuracy of the hypothesis, a definition of the sample (eligibility criteria) and the intervention, an estimation of the time required for the study, information on any missing data and, very importantly, provide evidence for the determination of the sample size for the subsequent clinical trial[16],[25].
Bioethical aspects and recording of protocol
The Declaration of Helsinki was developed by the World Medical Association in 1964 to provide ethical guidance for research involving human subjects, including such aspects as the duties of those conducting research involving human subjects. The importance of the research protocol, research involving vulnerable subjects, risk-benefit considerations, the importance of informed consent, the maintenance of confidentiality, and the reporting of findings to study participants. Although it is not legally binding in itself, many of the principles have entered legislation associated with research in most countries, thus it must be considered in the construction of any study with human beings. The Declaration has multiple revisions to date[17],[26] (https://www.wma.net/what-we-do/medical-ethics/declaration-of-helsinki/).
To initiate a clinical trial, it is necessary to bear in mind the basic principle stipulated by the Declaration of Helsinki[26] regarding the interventions to be studied. Considering potential benefits, risks, costs and effectiveness of any new intervention should be evaluated with respect to the best existing alternatives supported by evidence. There are several exceptions:
- Placebo, or no treatment, is acceptable in studies where an alternative intervention is unavailable and
- Where for scientifically sound and convincing methodological reasons, it is necessary to determine the efficacy and safety of an intervention using any intervention less effective than the best proven intervention, the use of a placebo or no intervention.
As with any research involving human subjects, clinical trials require a research protocol that must be reviewed and approved by a scientific-ethics committee[1],[27],[28], and registered in a trial’s registry prior to the enrollment of subjects[29],[30]. The purpose of registering the protocol is to detect any deviations after the study has been conducted, ensuring that authors report the outcomes they initially declared to be clinically relevant, thus avoiding selective outcome reporting[31]. This process provides transparency and visibility to clinical research, allowing those developing future clinical trials and systematic reviews of clinical trials to have an overview of ongoing research. All of this has been conceptualized in the Good Clinical Practice model, a standard for the design, conduct, performance, monitoring, auditing, recording, analysis and reporting of clinical trials, which safeguards the reliability of the results within a framework of investigative integrity and participant confidentiality[10].
At the end of the 20th century, several public registries for clinical trials originated. In the United States, the publicly funded Clinical Trials registry was created (http://clinicaltrials.gov), while in Europe the ISRCTN registry was established (http://isrctn.com), recognized by the World Health Organization and the International Committee of Medical Journal Editors (ICMJE) and supported by the Medical Research Council and the National Health System Research and Development Program, both of which are British organizations[32]. For its part, Cochrane has the Cochrane Central Register of Controlled Trials (https://www.cochranelibrary.com/central) and the World Health Organization has the International Clinical Trials Registry Platform (ICTRP) (https://www.who.int/ictrp/en/).
Essential components and procedures in clinical trials
Recruitment and randomization of participants
Recruitment of participants for a clinical trial is usually non-probabilistic sampling, which incorporates subjects that meet the eligibility criteria set out in the study protocol. This is also known as "convenience sampling." Subjects are then randomly allocated to receive an intervention or some comparator, which, as will be discussed below, is the best method for controlling selection and confounding biases[3],[33],[34]. This non-discretionary allocation of participants to study groups should be done strictly by chance, ensuring all participants have an equal chance of being included in any of the groups. As this process progresses, the groups tend to be more homogeneous, both in terms of confounding variables that are known and measured, as well as other variables associated with the outcome that were unknown or could not be measured. Randomization can be performed using a table of random numbers found in a statistical book, but usually computerized methods of randomization are used, such as computer-generated sequences. Particularly noteworthy is the concealment of the randomization sequence, which must be unknown to the researchers and clinical trial participants, such that it is not to possible to predict the group to which the next included participant will be assigned.
Estimating the number of participants to be randomized (sample size calculation) is a major part of randomization. How many participants are required to equalize confounding factors between intervention groups? More is not necessarily better, as people might be unnecessarily exposed to the risks of an intervention. However, if the number of patients randomized is less than the estimated sample size, results might be biased despite randomization[7].
Among the different types of randomization are simple randomization, where a unique sequence is generated by an entirely random procedure. In clinical trials with large sample sizes, simple randomization may generate a similar number of participants between groups, but in studies involving few participants it may result in unequal numbers in each group[35]. Another form of randomization is block randomization, which aims to ensure that the sizes of each group are similar[17]. Each block contains a similar number of participants assigned to each treatment, where the total number of participants has been predetermined by the researchers; the blocks are then randomly assigned to each group. The problem with block randomization is that the groups generated may be unequal with respect to certain variables of interest[35]. In light of this, stratified randomization is applied to ensure each group be assigned a similar number of participants with regard to characteristics of importance to the study, which must be identified by the researchers. In this type of randomization, different blocks of participants are configured with combinations of covariates that can influence the dependent variable to be explained (randomization according to prognostic factors). Then, a simple randomization is performed within each stratum to assign the subjects to one of the intervention groups. Therefore, it in order to carry out a stratified randomization, it is necessary to know the characteristics of each subject with precision[17],[35],[36] (Example 4). Finally, one method of randomization that has been used in clinical trials with a small sample size is adaptive randomization, in which a new participant is sequentially assigned to a particular intervention group, taking into account previous participant assignments as well as specific covariates. Adaptive randomization uses the minimization method, assessing the imbalance in sample size among multiple covariates, which could occur when applying simple randomization in a clinical trial with a reduced sample size[35],[37],[38].
Any assignment that does involve randomization, such as assignment by alternation does not qualify as random allocation[4]. Examples include assignment by alteration (the first participant is assigned to the intervention group, second participant to the control group, and so on), assignment according to the day of the week, assignment according to the initial letter of the first surname, among others.
Measurement of results
As stated, ideally masking methods are applied where possible to ensure participants are blind. This allows the emphasis placed by researchers on the measurement of results to be the same for all groups. Moreover, patients are not affected by the influence of knowing whether they are in the intervention group or not, with the aim to diminish subjective responses to treatment. Three related phenomena that may occur at the participant level are: the placebo effect[40],[41], the nocebo effect[40], and the Hawthorne effect[42],[43]. The first is associated with a reported improvement by the participant after receiving a substance with no active constituent, i.e., a placebo. In contrast, when a nocebo effect occurs, a substance or intervention without medical effects worsens a person's health status due to negative beliefs that the participant may have. The Hawthorne effect, also known as the "observer effect," occurs when participants in a clinical trial change their usual behavior knowing they are observed by a third party, affecting the point estimate of the intervention. These three phenomena are more prominent in randomized clinical trials that analyze outcomes reported by participants, i.e., where there is a larger subjective component (Example 5).
Data analysis
Once the outcomes of interest have been measured for the necessary duration, it is essential to be able to include all (or most) of the participants that were initially randomized in the primary statistical analysis. Participants may drop out of the study, although all reasonable measures should be taken by the investigators to avoid this. The analysis that includes all participant as part of the group to which they were assigned is known as an intention-to-treat analysis. The principle behind an intention-to-treat analysis is to preserve the benefit of randomization, or in other words, the balance of known and unknown prognostic and confounding factors, decreasing the probability of bias[45]. However, this approach is hampered when, having low adherence to the assigned treatment, not all participants are duly followed-up. In a per-protocol analysis, participants that did not adhere to the protocol are excluded from the analysis. This may seem reasonable at first, as it is of interest to know the effects of the intervention on the patients that actually received it. Nevertheless, it is recognized adherence to any treatment—how patients would take medicines in the real-world—that is usually lower than theoretically estimated, thus a per-protocol analysis is not representative of what happens in reality [46]. In turn, patients who are less adherent to therapy, even if it is a placebo, tend to have a worse prognosis than those who do adhere to it[47]. By excluding them from the treatment group and including them together with participants in the comparator arm of the analysis, the patients with the best prognosis are the only ones included in the treatment group, magnifying the estimated effect substantially[48] (Example 6).
Types of experimental studies on humans
This article has mostly dealt with the concepts related to the classical randomized clinical trial, the variants and the specifications of which are described below. However, there are other designs that also study the effects of an intervention on participants.
Classic randomized clinical trial
This clinical trial corresponds to phase III trial of the previous classification. Participants are randomized to receive a trial intervention or to a comparator. Randomization would ensure that the characteristics of the participants are distributed evenly across the groups; therefore, any significant differences in outcome between the groups can be attributed to the intervention and not to another unidentified factor. If the clinical trial seeks to evaluate an intervention under ideal (not every day) and rigorously controlled conditions, it is an explanatory clinical trial (efficacy study), whereas if its evaluation takes place in a context that emulates the circumstances of everyday life or clinical practice, it is called a pragmatic clinical trial[49]; in this sense, tools have been developed to assess the level of pragmatism of a clinical trial[50]. Another characterization relates to the objective of the study, as randomized clinical trials aim to demonstrate superiority, equivalence, or non-inferiority of one intervention over another[51]. In the first case, the aim is to prove that the treatment is better than another. In the second, the design and analysis will be oriented to determine therapeutic equivalence between two interventions, but where certain benefits such as fewer adverse effects, simpler use, or lower economic cost are expected. Non-inferiority studies are considered a special case of equivalence studies; these studies only analyze the novel intervention as not worse than an existing one[17].
By virtue of study design, we find the parallel randomized clinical trial, most frequently seen, where each group receives an intervention simultaneously. On the other hand, in crossover randomized clinical trials, each participant receives each intervention under study consecutively, so that each subject is his or her own comparator. Doing so essentially doubles the sample size, since subjects initially randomized to the intervention group will later receive the comparison and vice versa. A disadvantage of this design is the carryover phenomenon, where the effects of the first intervention can interfere with the effects of the second, so this type of trial is useful in interventions with effects that last a short period. However, it is desirable to space out both interventions temporarily (washout period), in order to decrease the likelihood that the first will interfere with the second[52],[53]. Therefore, when discontinuing the intervention, the condition of the subject who received it should be the same as before receiving it, since if it changes, the second intervention would be applied to a different participant than the one who received the first. For this reason, this design is limited to the study of chronic, time-resistant conditions[54] (Example 7).
Another existing design is the factorial clinical trial, which allows two or more research questions to be answered together. An example is when two or more interventions are evaluated separately and in combination against a control. Participants are randomized two or more times to one of the intervention groups, depending on the number of therapies to be studied. The great advantage is that it provides more information than a parallel design study. In addition, it permits the interaction between two treatments to be evaluated[56].
Another form of randomized clinical trial is one in which groups of participants or clusters (for example, health centers, geographic areas) are randomly assigned to an intervention or a comparison of an intervention. They are useful in the study of individuals with similar biological or psychosocial characteristics and for when the intervention analyzed has a group effect, such as non-pharmacological interventions, public policies[57],[58] or the effect of a vaccine[5].
Finally, there are randomized clinical trials of discontinuation. In these trials, patients who already receive some treatment are randomly assigned to continue their therapy or discontinue it and receive a placebo. Discontinuation designs are applied in chronic non-healing therapies whose effect is small[54].
Quasi-experimental studies
These are characterized by not applying a process of randomization of participants to the intervention or comparator group, so they are also known as non-randomized controlled trials. Although there are numerous examples of quasi-experimental types of studies in biomedical research, these designs come from the field of psychology and the social sciences[59], where due to certain circumstances the random assignment of subjects to experimental conditions is not possible. There are several types of quasi-experimental studies, including before-after or pretest-posttest designs and interrupted time series. We will highlight the before-after design, in which the same variable is measured before and after an intervention (each participant acts as his or her own control). Its results are useful when the effect is large, is consistently observed in most participants, and is therefore unlikely to be explained by chance[4] (Example 8).
Quasi-experimental studies tend to be simpler and involve a lower economic cost than randomized clinical trials, constituting an option when random assignment is impractical, when a bioethical impediment exists, or when the intervention needs to be performed under natural conditions. Its disadvantages are associated with a high susceptibility to confusion and selection bias. In addition, the placebo effect and the Hawthorne effect are especially relevant, which could be lessened if subjects are unaware of the intervention they are participating in[60],[61]. Statistical methods for paired groups should ideally be considered during data analysis, since repeated measurements will be made over time on the same group of subjects[62]. Therefore, interpretation and extrapolation of data obtained from non-randomized studies is complex[17].
Experimental studies with external or historical controls
They use as a control group subjects that do not come from the same population from which the sample was obtained (non-current sample), therefore, the comparison will be made from data of patients already published or from records of a health institution, that is, with people who have already received treatment and evaluation. A disadvantage of this methodology involves the differences that may exist between the intervention and control groups, making them not very comparable[4], since there are differences in the temporal context in which the treatments were applied, and the people who performed the intervention, among others[17].
Measures of association
Due to a prospective design, the measure of association to be used will be relative risk (RR), which is understood as the ratio of absolute risks between the group of individuals exposed to the intervention and those unexposed. If the relative risk is equal to 1, it is assumed that there is no association between the intervention and the outcome (that is, the associated confidence interval does not incorporate the value 1); if it is greater than 1, the intervention increases the probability of the outcome occurring compared to the comparator, while if it is less than 1, the intervention decreases the probability of the outcome occurring compared to the comparator. The interpretation will be different depending on the outcome measured, as it may be favorable or unfavorable; therefore, if the outcome studied is a decrease in depressive mood, a relative risk greater than one will be favorable, while if mortality is measured, a relative risk greater than one will be unfavorable[63],[64],[65].
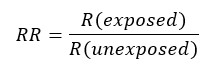
Other ways to express the magnitude of the effect is through differences. One of these is the absolute risk reduction (ARR), also known as attributable risk or risk reduction, which corresponds to the difference between the risks of the (unexposed) control group and the (exposed) intervention group—i.e. the risk reduction attributed to the intervention. If its result is negative, it is interpreted as an absolute increase in risk[63],[64],[65].
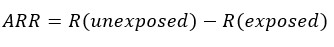
The relative risk reduction (RRR) corresponds to the difference in risk between the two groups with respect to the control group, or the quotient between the absolute risk reduction and the risk of the control group. If the result is negative, it is interpreted as a relative increase in risk. The relative risk reduction often overestimates the effects of treatment, so the absolute risk reduction should be reported where possible[63],[64],[65].
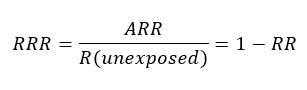
One measure derived from the absolute risk reduction is the number needed to treat (NNT), which quantifies the number of patients to be treated to prevent an event from occurring[63],[64],[65]; for example, if the number needed to treat with an antibiotic was 15, it is interpreted that 15 patients should be treated before one person recovers from an infectious condition.
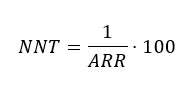
On the other hand, the number needed to harm corresponds to an index of the adverse events associated with a treatment, meaning the number of patients who should receive one treatment instead of another for an additional patient to present a harmful event. When the result of the number needed to treat is negative, it should be interpreted as the number needed to harm[63],[64],[65].
While absolute risk reduction, relative risk reduction, and number needed to treat provide information on the magnitude of the treatment effect, they are strongly related to the variability of the measured outcome parameter and the sample size, and should be reported along with confidence intervals that provide information on the precision of the findings[66].
Biases
A bias present in any type of research is the confounding bias, as it can never be completely eliminated. Nevertheless, clinical trials manage to reduce this significantly thanks to the process of randomization, which allows for the homogeneous distribution of known and unknown variables among the study groups. The following are some of the biases observed in clinical trials.
Selection bias
It occurs when the methods for selecting the sample from the population favor one group over another. It also appears when some relevant proportion of the target population is not included. This bias is controlled by an appropriate selection process of participants and their subsequent randomization[67].
Performance bias
There is a systematic difference between groups regarding the care and the follow-up provided. For example, performance bias is present when researchers keep closer track of patients assigned to the intervention under study. Standardization of procedures, adequate staff training and masking combat this source of systematic error[67],[68].
Detection or information bias
It happens when the outcome under study is "detected" differentially between groups, which can lead to different results. Detection bias is most prominent during the recording of subjective outcomes reported by participants (for example, analgesic response). Thus, if a researcher records the observed results in a way that supports his or her belief, a detection bias will emerge. This bias is controlled by blinding[67],[68].
Attrition bias
It occurs when there are systematic differences in the follow-up of clinical trial participants depending on the group they were assigned, resulting for example in loss of follow-up, which increases uncertainty in the results. This bias is controlled by performing an intention-to-treat analysis[67],[68].
Reporting bias
This bias is investigated during the presentation of the results, where a selective reporting of the outcomes of greatest interest or those that demonstrate the hypothesis under study may occur, which would increase the impact of the study and the probability of its publication. Reporting bias can be assessed by reviewing the initial clinical trial record and/or its published protocol and then comparing it with what has been reported[67],[68].
Reporting guidelines
Proper reporting of clinical trials is of great importance, as it allows the results and conclusions to be understood and interpreted, and ensures their reproducibility[69]. In order to standardize criteria for clinical trial reporting and facilitate critical reading and interpretation, the Consolidated Standards of Reporting Trials (CONSORT) initiative[70] was launched in the mid-1990s and is constantly being revised, updated and specialized. This proposal is made up of 25 items grouped into 6 domains: Title and Abstract, Introduction, Methodology, Results, Discussion and Other Information. A flow chart that presents numbers of participants per group, as they move through phases of the trial, should also be added, allowing a quick understanding of the most relevant aspects of the execution of the study. There are multiple versions of CONSORT adapted for specific interventions: CONSORT for non-inferiority studies[71], for pilot studies[72], for studies with herbal interventions[73], for pragmatic clinical trials[74], for clinical trials with psychological and social interventions[75], for single-participant (N-of-1) trials[76], for crossover clinical trials[53], among others. Briefly, when reading a published clinical trial, the following questions should be answered[5]:
- Is this a high-quality clinical trial that addresses an important question?
- Was randomization adequately performed?
- How complete was the follow-up? Was it similar among the groups in the study?
- Were positive and negative outcomes evaluated? Were evaluations blind?
- Are results applicable to clinical practice?
- Were patients adequately described?
- Was the intervention properly described?
- Was an intention-to-treat or per-protocol analysis performed?
In addition, the Transparent Reporting of Evaluations with Nonrandomized Designs (TREND) initiative[77] is aimed at reporting non-randomized intervention studies; it consists of 22 items grouped into 5 domains: Title and Abstract, Introduction, Methods, Results, and Discussion.
At the same time, scales such as the Jadad scale[78] and the scale developed by Cochrane[79] can be used to assess the quality of clinical trials.
Final considerations
Usually, the first table of results reported in clinical trial publications is "Table 1," which describes characteristics of interest of the participants of either group, both treated and comparator. It is normal to find p values of comparisons between groups for characteristics in Table 1 to support that the groups do not differ (p values greater than the significance level) and therefore these would not influence the results as confounding variables. However, this is not methodologically correct, since the research question and associated hypothesis test underlying the clinical trial relates to the difference between intervention and comparator, and not assessment of the frequency of a characteristic between groups. This has been emphasized by CONSORT[70],[80]. Some authors suggest a solution: if there is a difference greater than 10% between the frequency of the variables studied, the potential for confounding exists. In this instance, randomization might not have fully controlled potential confounding, and results should be interpreted with this residual effect in mind or, better yet, a multivariate regression model that incorporates the variable in question could be used to assess its effect as a confounder[7].
The prospective nature of clinical trials makes it possible to find causal relationships and to take steps to ensure the quality of the data obtained; nonetheless, it is necessary researchers allow for a latency time appropriate for the outcomes of interest. At this point, it is worth recognizing survival analysis, a statistical method mostly used in cohort studies[28]. This plots occurrence of an outcome over time for both groups, allowing comparison of the survival curves. Another statistical method applied in clinical trials is sequential analysis, which consists of conducting intermediate analyses to assess the need to continue or stop a trial depending on whether the hypothesis has been determined or evaluating the cost-benefit or risk-benefit balance, obeying pre-specified rules for continuation. Sequential analysis should be specified in the study protocol[81],[82].
Randomized clinical trials are sometimes criticized due to the low representativeness of participants resulting from non-probabilistic sampling, applying rigorous eligibility criteria. Likewise, the standardized implementation of the intervention might not resemble what happens in clinical practice, where interventions are likely less controlled and more heterogeneous. For this reason, external validity (extrapolation of the results) should be assessed with caution, as results may differ from those observed in widespread practice, where several factors involved were not controlled in the study[2].
Not all questions can be answered by an experimental study, such determination of risk factors for the development of lung neoplasia, where it would be unethical apply these to a group of people. In these cases, observational studies are an important option[28]. The study of adverse events associated with drug use should be reserved for observational studies or early phase experimental studies, but not in clinical experiments with humans. Thus, the analysis of rare adverse events remains a challenge, since the available studies are generally underpowered (due to an inadequate sample size to detect rare events), very similar comparison groups are required, and the analysis of the cause-effect relationship becomes complex[2].
Clinically relevant or significant outcomes need to be differentiated from subordinate, intermediate, indirect or surrogate outcomes[83]. Clinically relevant outcomes provide guidance on the use of an intervention and shed light on the effect of therapy, i.e. the direct measure of how a patient feels, lives and functions. In turn, surrogate outcomes may correspond to a laboratory parameter or a physical sign that does not directly measure the central clinical benefit of the intervention. Thus, the former are associated with the person themselves, while the latter are associated with the physiopathology of the disease. This consideration is important when reading randomized clinical trials, as it may be multiple positive outcomes are reported but none that are clinically relevant.
Finally, although randomized clinical trials are the cornerstone for studying the efficacy and safety of a therapy, a systematic review that meta-analyzes the results of multiple individual clinical trials that tested the same intervention represents an even higher level of evidence, as it provides a combined estimation of the effect of all the primary studies included[84]. The results of all clinical trials must be published to avoid publication bias, which occurs when investigators occult negative findings, or that may occur if journals are less inclined to accept a negative report. This leads the scientific community to observe a magnified effect of an intervention[6], which has a negative impact on society and is counterproductive to the social benefit of research.
 Full size
Full size 