Artículo de revisión
← vista completaPublicado el 24 de abril de 2023 | http://doi.org/10.5867/medwave.2023.03.2619
Efecto de la epigenética en la artritis reumatoide
Effect of epigenetics on rheumatoid arthritis
Abstract
Rheumatoid arthritis is an autoimmune and inflammatory disease that predominantly affects the diarthrodial joints. In this pathology, environmental or behavioral factors can act in synergy with genetic predisposition, accelerating the onset and severity of the disease. This link between the environment and the genome is mediated by epigenetic marks on deoxyribonucleic acid, including its methylation, histone modification, and noncoding ribonucleic acid-mediated regulation. Epigenetics can generate heritable phenotypic changes, which are not determined by modifications in the deoxyribonucleic acid sequence and are therefore reversible. Therefore, diet, medications and other environmental factors would have the ability to modulate them. The identification of a specific epigenetic dysregulation can offer a better understanding of the pathophysiology of the disease and positively influence the prevention, diagnosis and development of new therapeutic targets.
Main messages
- Rheumatoid arthritis is an autoimmune, inflammatory, and chronic condition that can cause disability.
- Therapeutic advances and clinimetry have improved the control of this disease.
- Knowledge of epigenetics has generated a series of elements, improving the conceptualization of the pathophysiological model of rheumatoid arthritis and integrating the interaction between genes and environment.
- In this narrative review, we evaluate the evidence of the advances in epigenetics and its effect on rheumatoid arthritis.
Introduction
The pathogenesis of rheumatoid arthritis involves cells of the innate immune system, such as macrophages, dendritic cells, and natural cytotoxic cells. In the adaptive immune system, we find T and B lymphocytes associated with the involvement of other non-immune cells, such as fibroblasts and endothelial cells [1].
Studies in first-degree relatives support the inheritance influence between 50% and 60%, inheritability is not modified by sex, and evidence suggests that seropositive rheumatoid arthritis with rheumatoid factor and/or anti-cyclic citrullinated peptide antibodies is more frequent in patients with a family history compared to those with seronegative rheumatoid arthritis [2].
Genome-wide association studies have characterized more than 100 genetic factors for rheumatoid arthritis, with the shared epitope being the best-known genetic risk factor, especially in the seropositive phenotype [3,4]. There are other non-antigen genes of the human leukocyte studied, i.e., protein tyrosine phosphatase non-receptor type 22 (PTPN22) [5,6], being the most susceptible molecule for rheumatoid arthritis in this group of molecules [5,6], also associated with risk for other autoimmune diseases such as type 1 diabetes mellitus, juvenile idiopathic arthritis, systemic lupus erythematosus, Sjogren’s syndrome, autoimmune thyroid disease, vitiligo and several forms of vasculitis. Likewise, polymorphism of the cytotoxic T lymphocyte antigen-4 (CTLA-4) gene [7,8] and dysregulation of the interleukin-23 receptor (IL-23R) have been analyzed as a predisposition factor for rheumatoid arthritis and other rheumatic conditions such as ankylosing spondylitis and systemic lupus erythematosus [9,10]. However, they do not fully explain the pathophysiological mechanisms of the disease due to the characteristics of polygenic inheritance patterns. It is precisely there where knowledge of epigenetics makes it possible to gather complementary elements that bring us closer to a better understanding of the disease.
Analyzing how these factors trigger an autoimmune response related to the onset of rheumatoid arthritis has led to a preclinical model involving the seropositive rheumatoid arthritis phenotype, characterized by the elevation of biomarkers, such as rheumatoid factor and/or anti-cyclic citrullinated peptide antibodies, in pre-disease phases. This suggests that the interaction of genes and the exposome [11,12] could interact before the first joint symptoms appear. However, this model is not comparable to seronegative phenotypes [13].
Epigenetics is a heritable and reversible phenomenon that compromises gene expression without generating changes in the primary sequence of deoxyribonucleic acid (DNA), and its three modifications are represented by:
-
DNA methylation in which the methyl group of S- adenosyl methionine is added to a cytosine base at CpG sites (DNA sites where the cytosine is followed by one at the guanine level).
-
Modification of histones by methylation and acetylation; chemical or other histone modifications; and covalent modifications of non-histone proteins.
-
A deregulated expression of micro-RNAs acting at the post-transcriptional level of genes [14,15].
These epigenetic mechanisms influence the phenotypic consequences of genetic variation. To explain the biological mechanism of a given pathology, it is necessary to characterize the relationship between genetic and epigenetic effects. These relationships can be described as mediation, in which genetic variation influences methylation, which then influences the phenotype or interaction (also called effect modification), in which the methylation effect differs according to genotype, or both [16].
This review aims to identify how specific epigenetic dysregulation can provide further insight into disease pathophysiology and affect the prevention, diagnosis, and therapeutic interventions.
Materials and methods
Search strategy and study selection
MEDLINE/PubMed, LILACS, and Cochrane databases were used. To broaden the search, we used Google Scholar and a manual search. The following terms were used: "Rheumatoid Arthritis" [Mesh] AND "Epigenetic" [Mesh], AND "DNA methylation. The inclusion criteria for this review were:
-
Works published in English and Spanish.
-
Clinical trials, cross-sectional studies, case-control studies, systematic reviews, and meta-analyses enrolling adult patients with rheumatoid arthritis according to the American College of Rheumatology and European League Against Rheumatism (ACR/EULAR) classification criteria.
-
Papers published from 2018 to 2022.
The exclusion criteria were:
-
Studies unrelated to this topic.
-
Editorials, narrative reviews, congress abstracts;
-
Articles without access.
-
Insufficient data.
-
Duplicate publications.
Epigenetic perspectives in rheumatoid arthritis
The etiology of rheumatoid arthritis, as in other autoimmune diseases, is multifactorial, with genetic, environmental, and endogenous factors playing an important role. All these factors may modify the epigenetic landscape of cells that are relevant in the pathogenesis of rheumatoid arthritis. This suggests that epigenetics may be a common interface in which risk factors, individually or together, act in the development of the disease. Numerous studies demonstrate differences in the epigenome of relevant cells in patients with rheumatoid arthritis compared to healthy individuals. The chronic inflammatory environment in rheumatoid arthritis may profoundly affect cell epigenetics provoking long-lasting changes in cellular phenotypes, obstructing the resolution of rheumatoid arthritis, and potentiating its chronicity. Elucidation of how rheumatoid arthritis risk factors change its epigenetics and how disease-related epigenetic alterations modify rheumatoid arthritis biology is the next step toward a better understanding of this pathology [17].
Epigenetic mechanisms, such as DNA methylation, chromatin remodeling, and non-coding ribonucleic acids (RNA), have been identified as crucial regulators in cellular immunity. This is due to their mechanisms for modulating gene expression and transcription in specific cells and tissues. DNA methylation is one of the most studied epigenetic marks, and it influences a wide variety of biological processes, such as transcriptional repression, reversible promoter silencing, and chromosomal instability. One of the crucial functions of DNA methylation is the maintenance of T-cell regulation. Therefore, DNA methylation plays a critical role in numerous autoimmune diseases by altering gene expression profiles. Epigenetic mechanisms involved in rheumatoid arthritis include altered methylation states in T and B cells and synovial fibroblasts [18].
Several lines of evidence suggest that epigenetic changes, including methylation and changes in microRNAs and long non-coding RNAs, happen before treatment or are induced by treatment and/or aging and may play an important role in patients with persistent inflammatory refractory rheumatoid arthritis. The epigenetic signature of rheumatoid arthritis may also change due to aging or environmental factors. In addition, initial successful treatment regimens in patients with rheumatoid arthritis can subsequently fail, suggesting a change in disease mediators in these individuals [19].
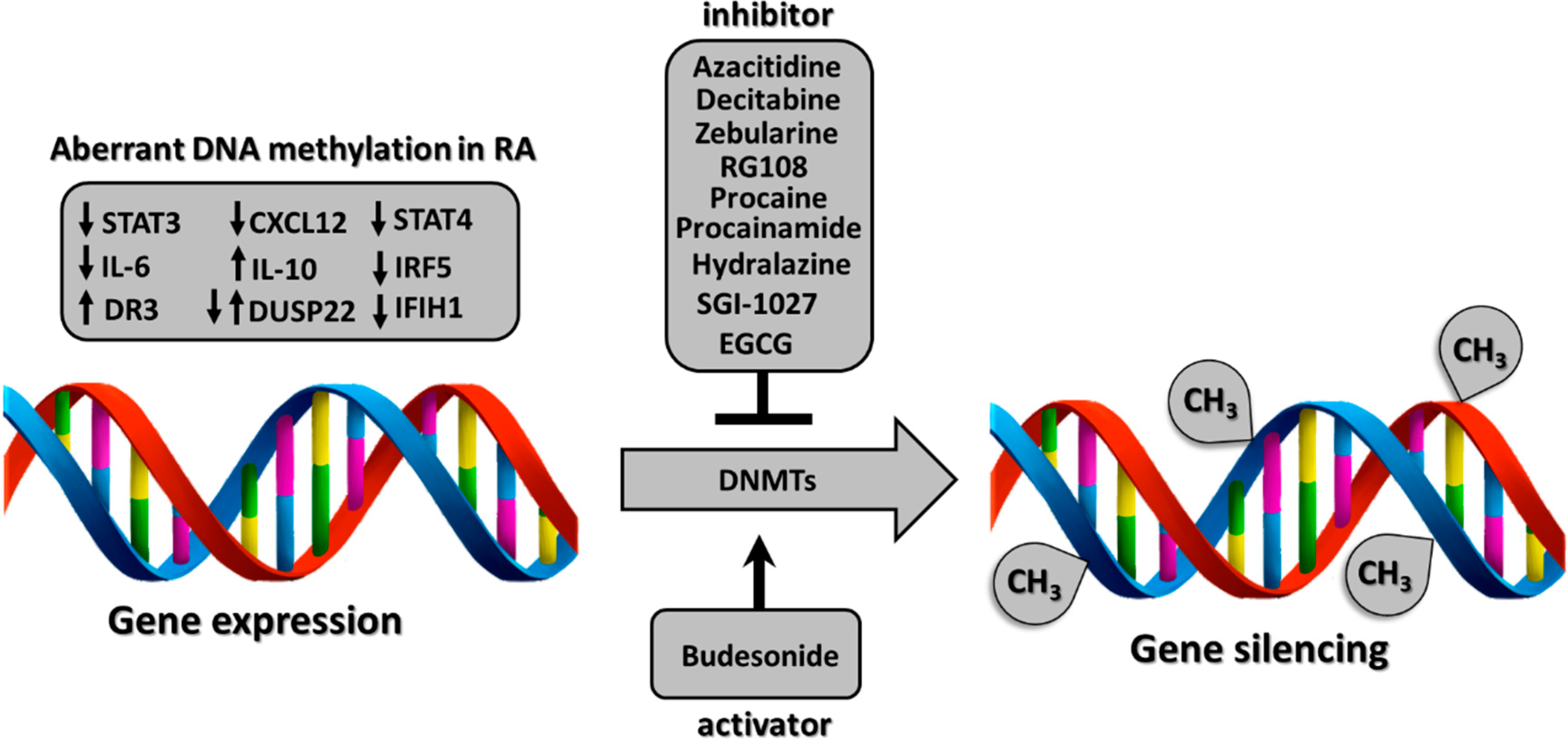
Some studies have shown that therapy with conventional synthetic disease-modifying antirheumatic drugs, biologics, and/or corticosteroids can influence DNA methylation status. Such methylation is exerted by a family of DNA methyltransferases, which are inhibited or activated by various epigenetic drugs and consequently modulate gene expression [20] (Figure 1).
Potential mechanism of differentially methylated genes in rheumatoid arthritis (RA) regulated by various epigenetic drugs. multiple epigenetic drugs.
DNA methylation is exerted by DNA methyltransferases. DNA methyltransferases are influenced by epigenetic drugs that inhibit or activate them, leading to gene expression modulation. The down-arrow indicates hypomethylated genes (STA3, interleukin-6, CXCL12, DUSP22, STAT4, IRF5, IFIH1) and the up-arrow indicates hypermethylated genes (DR3, interleukin-10, DUSP22).
In addition, miRNAs have been shown to play an important role in rheumatoid arthritis. Among them is miR-146a, proposed as a key regulator of the immune system by controlling cytokine secretion, B-cell function, and nuclear factor-kβ signaling, among others. Elevated levels of miR-146a have been associated with interleukin-17 expression in peripheral blood mononuclear cells and the synovial membrane of patients with rheumatoid arthritis. Other relevant microRNAs include miR-155, which produces up-regulation of tumor necrosis factor-α and interleukin-1β in peripheral blood mononuclear cells of individuals with rheumatoid arthritis; miR-346, which controls tumor necrosis factor-β synthesis; miR-23b, which suppresses autoimmune inflammation associated with interleukin-17; and miR-126a, which promotes inhibition of DNA methyltransferases, producing hypomethylation of interleukin-17-associated autoimmune inflammation; miR-23b, which suppresses interleukin-17-associated autoimmune inflammation; and miR-126a, which promotes inhibition of DNA methyltransferases, producing CD70 hypomethylation in B cells and thereby contributing to the autoimmune response [21].
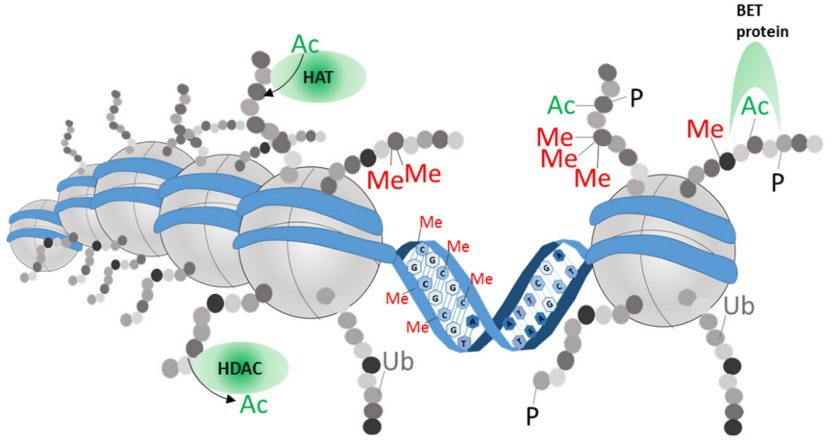
Finally, histone-modifying enzymes are epigenetic regulators involved in controlling inflammatory processes, including stromal and immune cell activation, survival, and proliferation. Histone acetyltransferases acetylate lysine residues on histone tails, whereas histone deacetylases counteract histone acetyltransferase activity by deacetylating histone proteins. The delicate balance between the acetylated and deacetylated states of chromatin affects both the overall structure and metabolism of chromatin, leading to gene activation or repression. In addition, histone acetyltransferases and histone deacetylases can also affect the acetylation state of non-histone proteins, thereby regulating signaling proteins and transcription factors to influence gene expression and cellular function; as a consequence, histone deacetylase function could be essential for the development and perpetuation of chronic inflammatory diseases, such as rheumatoid arthritis [22] (Figure 2).
Epigenetic modifications.
DNA (blue) is coiled around histones (gray). Amino acid modifications to the protruding histone tails can influence DNA density and recruit repressor or transcriptional complexes. Histone marks are positioned, read, and removed by specialized enzymes. For example, histone acetyltransferases (HATs) and histone deacetylases (HDACs) are shown. Bromo and extra-terminal domain (BET) family proteins bind to acetylated lysine on histone tails, after which they can promote transcription and recruitment of epigenetic enzymes to further stabilize the open chromatin state at a specific locus. Ac: acetylation, Me: methylation, P: phosphorylation, Ub: ubiquitination.
Epigenetics in rheumatoid arthritis
Epigenetics represents an important area for understanding some factors that contribute to the risk of developing rheumatoid arthritis, evidenced by the results of some studies on how epigenetic modifications can affect gene expression in this disease entity.
Tseng et al. conducted a study in a cohort of patients with rheumatoid arthritis and healthy controls on the effect of methylation of DNA coding for programmed cell death molecule 1, observing in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs, an increase in the expression of programmed cell death molecule 1 and hypomethylation of this molecule compared to controls. In addition, the expression of programmed cell death molecule 1 is impaired in cells carrying the T allele of the rs41386349 polymorphism compared to cells carrying the C allele. This defective expression of programmed cell death molecule 1 can lead to aberrant activation of humoral immune responses and increased autoantibodies. Consequently, this implies increased seropositivity (rheumatoid factor) [24].
A cross-sectional study demonstrated that peptidyl-arginine deiminase contributes to the pathogenesis of rheumatoid arthritis by inhibiting fibroblast-like synoviocyte apoptosis. Decreased peptidyl-arginine deiminase expression increases fibroblast-like synoviocyte apoptosis in patients with rheumatoid arthritis. In contrast, positive upregulation of peptidyl-arginine deiminase could decrease the apoptosis of fibroblast-like synoviocytes by disrupting modifications of the arginine residue of histone H3, which suppresses p21 transcription, providing new insight into the anti-apoptotic role of peptidyl-arginine deiminase in the development of rheumatoid arthritis and its implications for epigenetic mechanisms [25].
Ham et al. evaluated DNA methylation profiles of synoviocytes isolated from joint replacement surgery of patients with rheumatoid arthritis and osteoarthrosis. Among the 28 differentially expressed microRNAs (DEmiRs) between rheumatoid arthritis and osteoarthrosis, 27 microRNAs showed significant correlations with DNA methylation at the CpG level, suggesting that individual CpGs may have a potential role in microRNA regulation [26].
In a meta-analysis, which included 22 case-control studies (n = 10 489), we analyzed whether polymorphisms in microRNA genes were associated with rheumatoid arthritis. In this meta-analysis, the association of the rs2910164 (G/C) miR-146a polymorphism with rheumatoid arthritis was not found, whereas the association with the rs3746444 (T/C) miR-499 polymorphism was found, particularly in Caucasian populations [27].
Payet et al. performed a meta-analysis to evaluate microRNA expression, DNA methylation, and histone modifications in rheumatoid arthritis, observing that in human samples, miR-155, miR-146a, and miR-150 expressions decreased. In contrast, miR-410-3p expression increased, miR-146a decreased expression of the cytokine interleukin-17 and evidenced that DNA appears hypomethylated [28].
Epigenetic mechanisms as biomarkers
The polygenic complexity and the phenotypic differences presented by patients with rheumatoid arthritis drive the need to address other areas that contribute to preventing the onset of this pathology. Likewise, it is necessary to improve the clinical control of the disease, for which we cite some studies that analyze some biomarkers of importance.
One study, which included 17 synovial tissue samples from three cohorts, evaluated differences in DNA methylation and gene expression and their relationship to rheumatoid arthritis pathogenesis. The first cohort included individuals who had arthralgia and/or a family history of rheumatoid arthritis, a positive rheumatoid factor immunoglobulin M (IgM-RF), and/or anti-citrullinated protein antibodies but no proven disease. The second cohort included individuals with early arthritis, with a disease duration of less than one year, and who had not received prior treatment with disease-modifying antirheumatic drugs. The third cohort included patients with established rheumatoid arthritis, with a disease duration of more than one year, with at least one inflamed joint and active treatment. The analysis showed that the synovium of patients with a higher number of inflamed joints (equal to or greater than nine) was different in DNA methylation and gene expression level compared to the synovium of patients with a lower number of inflamed joints. The most significant difference was found in the methylation of the MIRLET7B and MIR10B promoter regions. At the transcript level, they were CCL13 and CLEC10A. Differential methylation in the vicinity of the promoter was also evident for CCL13, CXCL11, and CXCL9. Finally, differential transcript expression from ITGB2 and LCP2 was highlighted, with multiple regions of differential methylation observed around both genes. These results support the role of epigenetic/transcriptional processes with increased joint involvement [29].
Bae et al. evaluated, through a meta-analysis, the relationship between miR-146a levels and rheumatoid arthritis and its correlation with disease activity. The analysis showed that miR-146a levels in synovial tissue/fluid were significantly higher in patients with rheumatoid arthritis than in the group with osteoarthrosis. In addition, they observed a positive correlation between miR-146a and levels of erythrocyte sedimentation rate [30].
Ayeldeen et al. evaluated in a case-control study the use of microRNA-146a and microRNA-499 expression in the diagnosis of patients with rheumatoid arthritis. It showed that microRNA-146a (rs2710164) and microRNA-499 (rs3746444) polymorphisms were associated with increased susceptibility to rheumatoid arthritis [31].
Another case-control study evaluated FCER1G gene methylation and miR-17 family members as epigenetic markers associated with rheumatoid arthritis, disease activity, and interleukin-6 expression. The findings showed lower methylation of FCER1G and miR-106b in rheumatoid arthritis patients compared to controls but not among groups of patients with highly active rheumatoid arthritis and disease remission. Furthermore, they observed a correlation between interleukin-6 levels and FCER1G methylation [32].
Zhang et al. analyzed the association between single nucleotide polymorphisms in the microRNA-146a (miR-146a) gene and rheumatoid arthritis susceptibility. The analysis showed that the T allele of the functional single nucleotide polymorphism rs2431697 increases the risk of rheumatoid arthritis. The effect size of the single nucleotide polymorphism is larger in Asians than Europeans. Finally, they found significant heterogeneity in the trans-ethnic meta-analysis but not in the Asian meta-analysis [33].
In the serum determination of ADAMTSL2 and LRPAP1 gene methylation levels of patients with rheumatoid arthritis who were divided according to disease activity and compared to the control group, hypomethylation of ADAMTSL2 was observed in the group with DAS-28 (Disease Activity Score-28) and high disease activity compared to patients in remission, as well as in the control group. For the LRPAP1 gene, there was hypermethylation in the DAS-28 and the control groups compared to patients in remission [34].
Epigenetics and therapeutics
The advances in personalized medicine in rheumatology represent a challenge in terms of identifying biomarkers capable of defining patient groups that may respond differently to conventional synthetic disease-modifying antirheumatic drugs and/or biologics. The same is true for the need to develop epigenetic drug targets.
Gosselt et al. developed a randomized clinical trial to analyze the association between global DNA hydroxymethylation before, during, and after three months of methotrexate therapy and its relationship with changes in disease activity in patients with early rheumatoid arthritis. In that trial, they showed that higher baseline global DNA methylation is associated with a lack of clinical response, determined at three months of methotrexate therapy. In addition, mean global DNA methylation did not change during treatment with this drug, and global DNA methylation at three months or more was not associated with clinical efficacy [35].
Nair et al. investigated differential DNA methylation as a candidate biomarker of response to methotrexate in a multicentered observational study. This study demonstrated that four CpGs (cg04334751, cg23700278, cg27427581, cg26764200) exhibited methylation changes that predicted improvement in swollen, painful joints, decreased C-reactive protein and DAS-28 in patients with rheumatoid arthritis who initiated methotrexate treatment for the first time, at four weeks and six months follow-up. These results represent an advance in current practice by contributing to a personalized medicine strategy that may allow a change in therapy at four weeks [36].
Kim et al. performed an observational study where they treated epigenetically modified human mesenchymal stromal cells (epi-hMSCs) from patients with rheumatoid arthritis with a combination of hypomethylating agents such as 5-azacitidine or 5-aza-2′-deoxycytidine and histone deacetylase inhibitors such as trichostatin A or valproic acid, testing 36 treatment combinations. For this purpose, indolamine 2,3-dioxygenase and interleukin-10 expression levels in human mesenchymal stroma treated for 72 hours were used to select optimal drug combinations. It was found that treatment with the combination of a hypomethylating agent and a histone deacetylase inhibitor increased the immunomodulatory properties of human mesenchymal stroma, showing that an optimal combination of hypomethylating agents and histone deacetylase inhibitors can enhance the immunomodulatory potential of human mesenchymal stroma, which may be useful for the treatment of rheumatoid arthritis [37].
On the other hand, the Rotterdam cohort of early arthritis patients did not identify differentially methylated positions and regions in the genome in relation to changes in the activity scale measured with DAS-28 during the first three months of treatment. Larger studies are required to prove or rule out the use of DNA methylation sites as a predictive marker for response to methotrexate [38].
Guderud et al. in an epigenome-wide cohort and association study, which aimed to analyze rheumatoid arthritis relevant T cells (virgin and memory CD4+ T cells) from two different rheumatoid arthritis cohorts: rheumatoid arthritis patients without prior treatment with disease-modifying drugs and active disease, and rheumatoid arthritis patients treated with methotrexate who had been in remission for more than 12 months. It demonstrated more methylated positions on memory CD4+ T cells than on virgin CD4+ T cells (904 vs. 19 methylated positions) in patients with rheumatoid arthritis compared to controls. Most methylated positions identified in newly diagnosed, previously untreated rheumatoid arthritis patients with disease-modifying antirheumatic drugs and active disease showed increased DNA methylation (39 differentially methylated positions). In contrast, most methylated positions identified in rheumatoid arthritis patients treated with methotrexate in remission showed decreased DNA methylation. In addition, they found that approximately one-third of the 101 known rheumatoid arthritis risk loci overlapped (±500 kb) with the methylated positions. In particular, the introns of the UBASH3A gene harbor both the rheumatoid arthritis risk single nucleotide polymorphisms and two differentially methylated positions in CD4+ memory T cells [39].
Mortazavi et al. conducted a randomized clinical trial to evaluate the drug β-d-mannuronic acid and its effect on the expression of miR-146a, its targets (IRAK1, TRAF6), and the nuclear transcription factor, nuclear factor-κB, in patients with rheumatoid arthritis. After 12 weeks of treatment with β-d-mannuronic acid, they observed a decrease in gene expression of miR-146a, IRAK1, TRAF6, and nuclear factor-κB, as well as in serum levels of interleukin-6 and tumor necrosis factor-α [40].
Paoletti et al. analyzed membrane tumor necrosis factor expression in blood monocytes, macrophage polarization, miR-155 expression, and anti-tumor necrosis factor effect in patients with rheumatoid arthritis. The analysis showed that patients with rheumatoid arthritis had specific monocyte/macrophage abnormalities, that increased membrane tumor necrosis factor in monocytes was associated with disease activity, and that maturation of monocytes into anti-inflammatory M2-like macrophages was impaired. Moreover, increased expression of miR-155 in monocytes/macrophages could be responsible for this monocyte polarization defect, which an anti-tumor necrosis factor monoclonal antibody could partially reverse and not by the soluble receptor. Therefore, it can be suggested that an antagonist of miR-155 on monocytes/macrophages may represent a new therapeutic strategy in rheumatoid arthritis [41].
In addition, two randomized clinical trials demonstrated the effect of the drugs on the expression of microRNAs. A first trial showed, in patients with early rheumatoid arthritis, elevated levels of miR-27a-3p were associated with remission after 12 months of treatment with adalimumab and methotrexate. In this same trial, they demonstrated that miR-16-5p and miR-22-3p might be biomarkers of disease activity and predictors of response to methotrexate treatment [42]. The second trial demonstrated that patients treated with the drug β-d-mannuronic acid showed a decrease in miR-155, an increase in SOCS1, SHIP1, and a decrease in transcription factor nuclear factor-κB, generating a decrease in disease activity quantified by the DAS 28 scale and improvement in quality of life measured by the HAQ scale [43].
In a systematic review on the utility of microRNAs as biomarkers of response to treatment with methotrexate, tumor necrosis factor inhibitors (anti-TNF), and rituximab in patients with rheumatoid arthritis, suggesting that the most polyvalent microRNAs were miR-146a, which predicted response to methotrexate and tumor necrosis factor inhibitors, and miR-125b, which predicted response to infliximab and rituximab [44].
Discussion
It was determined that the most relevant microRNAs as susceptibility factors in rheumatoid arthritis were represented by microRNA-146a and microRNA-499. It is important to analyze the role of these molecules in different ethnic populations and to correlate them with serological and clinical biomarkers. They were also related to disease activity and possible predictors of treatment response.
The action of methotrexate on hydroxymethylation in patients with rheumatoid arthritis has been evidenced. It is important to consider research work evaluating the effect of different drug doses on methylation. We have noted the description of experimental drugs, such as β-d-mannuronic acid, and others involved in epigenetics-related processes. However, the current scientific support for their use in humans is incomplete, requiring studies that address the therapeutic impact on epigenetic mechanisms in patients with rheumatoid arthritis.
We were able to review that there are inconclusive studies on certain epigenetic mechanisms, which corroborates the complexity underlying the understanding of rheumatoid arthritis. This is because it is a chronic, polygenic disease that, in order to be clinically expressed, requires interaction with the exposome, understood as the conjunction of specific external exposure (environmental pollution, tobacco, etc.), general external exposure (socioeconomic status, education, etc.), and internal exposure (inflammation, oxidative stress, microbiome, etc.) [45]. As a correlation of this last element, it is necessary to point out how these effects of the external environment induce changes in organisms in the cellular redox state and, even more specifically, in their nuclear redox compartment. All of these would induce the modulation of epigenetic regulation in cells by post-translational modification of these proteins, such as histone nitrosylation, carbonylation, or glutathionylation [46]. Therefore, future research initiatives should incorporate these elements for a more integrative analysis.
Conclusions
Epigenetics represents a component within the pathophysiological complexity of rheumatoid arthritis in its prevention, diagnosis, and therapy. Consequently, its progress and relationship with the other integrative factors of the disease will allow a better understanding of this pathology.
