Artículo de revisión
← vista completaPublicado el 8 de enero de 2025 | http://doi.org/10.5867/medwave.2025.01.2994
Aspectos genéticos y neurobiológicos de la psicosis en el trastorno neurocognitivo mayor
Genetic and neurobiological aspects of psychosis in major neurocognitive disorder
Abstract
Psychiatric symptoms are frequent in neurocognitive disorders and dementias. Psychotic symptoms, mainly hallucinations and delusions, may appear in up to 50% of cases, influencing morbidity and mortality. Genetic, neurobiological, and environmental factors are involved in their onset. We conducted a narrative review of primary articles developed in humans that analyzed the genetic and neurobiological basis of psychosis in dementias. Evidence suggests that there are genetic risk variants for presenting psychosis in dementia. How genetic variants are related to schizophrenia, dementia, and other neurodegenerative disorders is under discussion. Candidate gene studies have found and genetic variants are associated with psychosis in dementia while genome-wide association studies have shown variants located in y . Epigenetic studies are scarce but have detected differences in the methylome of people with dementia and psychosis. On the other hand, alterations of the cholinergic, serotonergic, dopaminergic, and gabaergic neurotransmitter systems and the excitatory-inhibitory balance have been described in dementia. From a functional and anatomical point of view, there are alterations in several regions, mainly in the frontal area and other sensory processing and integration areas. Finally, we describe the influence of cognitive alterations in the genesis and maintenance of delusions and discuss the phenomenological overlap with confabulations. Multiple genetic, neurobiological, structural, and cognitive factors influence the occurrence of delusions and hallucinations in persons with dementia. Further research is needed to understand the pathophysiology of psychosis in dementias. This approach would support the understanding of psychosis as a transdiagnostic entity.
Main messages
- Psychotic symptoms are common in neurocognitive disorders and are associated with greater comorbidity and worse prognosis. Their temporal onset in the development of dementia is unclear, and multiple biological factors are involved in the genesis of psychotic symptoms in neurocognitive disorders.
- Genome-wide association studies (GWAS) have found genetic risk variants in the ENPP6 and SUMF1 genes, while epigenetic research regarding this topic is scarce.
- There is no consistent evidence on the relationship between the genetic basis of chronic psychosis and psychosis in dementia.
- Among the neurobiological factors, alterations in several neurotransmitter systems and brain morphofunctional changes are associated with neurodegeneration, especially in the prefrontal, frontostriatal, and limbic regions.
Introduction
Mild neurocognitive disorder and major neurocognitive disorder, or dementia, are chronic and often form a continuum [1]. The prevalence of dementia in people over 65 years of age is estimated at 5%, while it ranges between 20 and 40% in people over 85 years of age. About 50 million people are affected worldwide, reaching 139 million by 2050 [2]. The most common cause of dementia is Alzheimer’s disease (70% of cases) [3], followed by vascular dementia, frontotemporal dementia, dementia associated with Parkinson’s disease, and Lewy body dementia. However, epidemiological indicators fluctuate according to the samples studied and the age of onset [4]. Other types of dementia are those related to traumatic brain injury, mixed dementias, and rare-cause dementias [4]. Their clinical manifestations are diverse, including cognitive and behavioral disturbances. However, symptoms from many other areas coexist, such as psychotic symptoms. In this spectrum, hallucinations and delusions are frequent, which mainly consist of prejudice, theft, and false recognition. Unlike most purely psychiatric conditions, dementias may present more frequently with visual hallucinations.
Delusions are seen in about 65% of those with dementia, regardless of the cause [5]. Psychotic symptoms are most frequent in Lewy body dementia, followed by dementia associated with Parkinson’s disease, vascular dementia, and frontotemporal dementia [6]. Carriers of psychiatric disorders, particularly of the psychotic spectrum, present more frequently with dementia and vice versa [7]. In Alzheimer’s disease, psychotic manifestations can be evidenced in up to 50% of patients [8], being associated with worse cognitive alterations, aggressiveness, depression, and mortality [9]. Specifically, visual hallucinations are described in about half of the patients with Parkinson’s disease and most patients with Lewy’s disease [10].
Psychotic symptoms may precede cognitive impairment in older adults, in whom a delusional disorder or late-onset schizophrenia may be diagnosed. This fact complicates the clinical scenario. Psychosis may also occur in mild neurocognitive disorders, which often prelude dementia [11]. Although evidence is scarce, this suggests that psychosis comorbid to mild neurocognitive disorders constitutes part of a single neurodegenerative process, supporting a syndromic approach to psychosis as part of dementia [12].
Late-onset psychoses could originate from neurodegenerative processes distinct from typical forms of early-onset psychosis. They could also be considered non-cognitive symptoms of neurodegenerative processes [12]. Specifically, the pathophysiology of Alzheimer’s disease, such as neurofibrillary tangles and neuritic plaques, as well as vascular lesions, may play a role in psychotic symptoms [13]. This means that psychosis in Alzheimer’s disease may be mediated by different mechanisms that converge in a common pathway.
Dementia has a heritability of about 30% [14], and polygenic forms are much more frequent than those with Mendelian inheritance. This implies that most cases of dementia involve multiple genetic risk variants with additive and small effects. In this sense, if different forms of dementia have a different risk of psychotic symptomatology, it could be argued that the genetic architecture differs between neurocognitive disorders, which in turn generates a differential risk of presenting psychosis. Knowledge of the genetic makeup of dementia has been advanced by the development of extensive genome-wide association studies (GWAS). In these studies, different significant polymorphisms have been found, and their association with the known genetic architecture of psychosis has been analyzed.
In this review, we describe the main findings regarding genetic and neurobiological factors related to psychosis in dementia based on the evidence provided by primary studies in humans. We initially address genetic studies performed on candidate genes and then on genome-wide association studies. Then, we discuss the main neurobiological aspects, including the cholinergic, serotonergic, dopaminergic, and glutamatergic neurotransmitter systems and the most replicated morphofunctional findings. Finally, we describe some cognitive models proposed to explain the occurrence of false recognition delusions.
Genetic evidence
Alzheimer’s disease presents familial aggregation [15], with heritability initially estimated at 61% [16] and later specified in a range of 18 to 31% [14]. The latter study verified the first two significant loci for psychosis in Alzheimer’s disease, ENPP6 and SUMF1, whose role in the pathophysiology of the disease is not known. ENPP6 is related to oligodendrocyte metabolism, whereas SUMF1 is involved in lysosomal physiology [14]. DeMichele-Sweet and Sweet [17] proposed three possible genetic models. In all of them, the presence of genetic variants linked to psychosis located in genes such as COMT (catechol-O-methyltransferase), NRG1 (neuregulin-1), and CHRNA7 (nicotinic α7 acetylcholine receptor) play a central role. Genetic variants of COMT interact with some polymorphisms associated with Alzheimer’s disease, such as the ε4 allele of APOE (apolipoprotein E). NRG1 variants modify the course of other neurodegenerative diseases, ending in a similar phenotype. And CHRNA7 variants interact with others involved in neurodevelopmental diseases, such as schizophrenia, to conclude in the same phenotype.
In a subsequent study published in 2018 [18], the same research group analyzed a new cohort of 2876 people with Alzheimer’s disease. Their findings were replicated in a second cohort of 2194 people with Alzheimer’s disease, with and without psychosis. All participants were genotyped using an array that included single nucleotide polymorphisms (SNPs) with known significance for Alzheimer’s disease with psychosis and other SNPs associated with schizophrenia risk (1574 single nucleotide polymorphisms). It was concluded that the Alzheimer’s disease phenotype with psychosis had an inverse genetic association with the risk of developing schizophrenia since the polymorphisms associated with schizophrenia had a protective role for developing psychosis in Alzheimer’s disease. These results were corroborated in both cohorts. The authors argue that the polygenic risk of schizophrenia would be shared with disorders such as autism and bipolar disorder, which in their biological and clinical characteristics differ significantly from neurodegenerative diseases such as Alzheimer’s disease. They point out that some risk variants for schizophrenia related to neurodevelopmental alterations could have a protective role in Alzheimer’s disease. Indeed, the activation of microglia-mediated activity of specific receptors could be deleterious during neurodevelopment by altering the synaptic structure, thereby increasing the risk of schizophrenia. However, it would be protective for neurodegenerative disease, as it would augment β-amyloid peptide clearance.
DeMichele-Sweet et al. (2018) [18] performed a meta-analysis, finding no polymorphisms with a significant level of statistical association for a genome-wide scan study. However, three SNPs had the highest magnitude associations: rs300215, rs6859958 and rs999581. All of them are located at the RP11-541P9.3 locus, whose function is unknown but could modulate its function due to proximity to CCNG1 [18]. CCNG1 encodes for cyclin G1, a protein linked to cell cycle regulation through its interaction with proteins with kinase activity, i.e., capable of phosphorylation. Therefore, it could be involved in psychosis in Alzheimer’s disease since the finding of phosphorylated τ aggregates is a central phenomenon in this condition [9]. In a second genome-wide association study published in 2021 [14], DeMichele-Sweet et al. found an inverse, but not significant, genetic correlation between psychosis in Alzheimer’s disease and schizophrenia.
On the other hand, some authors point out that Alzheimer’s disease may share some genetic risk with schizophrenia. This was shown by a study that found that the polygenic risk score for schizophrenia was associated with the presence of psychotic symptoms in people with Alzheimer’s disease, especially in the case of delusions [19]. This risk could be given by some genetic variants that have been more consistently associated with the presence of schizophrenia. An example is the SS genotype for the polymorphic position BalI (biallelic, with G and S alleles), located in the gene encoding the dopamine D3 receptor (DRD3). However, one investigation did not find an association between the different BalI variants and the presence of psychotic symptoms in Alzheimer’s carriers [20].
The presence of neurofibrillary tangles would be linked to psychotic symptoms in Alzheimer’s disease. For this reason, the study of the MAPT gene, which codes for the microtubule-associated protein τ, has become relevant. Creese et al. [21] published in 2014 an analysis of data provided by two clinical trials on memantine, verifying that the H2 allele of the MAPT haplotype was associated with a 5.4-fold increased risk of increased severity of hallucinations, even after adjusting for the severity of dementia, the use of memantine, and antipsychotics. For its part, the gene encoding for the serotonin transporter (SLC6A4) presents the polymorphic promoter region 5-HTTLPR, with alleles known as "short" (S) and "long" (L). Both have been differentially linked to several neuropsychiatric conditions, including dementia.
In particular, Lewy body dementia and Parkinson’s disease-associated dementia share some clinical and pathophysiological features. In this regard, a study compared psychotic symptomatology in 97 participants with one of these dementias with 90 healthy participants [22]. The results indicated that the LL genotype of 5-HTTLPR was significantly associated with the risk of delusions but not hallucinations. Similarly, Borroni et al. [23] analyzed the implications of 5-HTTLPR variants on the risk of psychosis in Alzheimer’s disease, finding a significant association. However, unlike the previous group, the risk was associated with carrying the S allele of 5-HTTLPR. This study also found that the high-activity allele of the catechol-O-methyltransferase enzyme was linked to an increased risk of psychosis in Alzheimer’s disease. Interestingly, when combining the effect of the variants in both genes, the risk was increased fivefold compared to those who did not carry the variants. In this sample, the apolipoprotein E genotype did not impact the risk of psychosis.
Scasselatti et al. [24] studied various psychological symptoms in an Italian cohort of 362 patients with Alzheimer’s disease. In their work, they found that those who did not carry the ε4 allele of apolipoprotein E had an increased risk of developing psychosis. Together, they concluded that CC homozygotes for the single nucleotide polymorphism C677T (rs1801133) located in the methylenetetrahydrofolate reductase (MTHFR) gene showed an increased risk of delirium. This variant has been associated with other neuropsychiatric syndromes, such as schizophrenia. The authors found no significant associations between catechol-O-methyltransferase gene variants and phenotype. The genome-wide association study by DeMichele-Sweet et al. (2021) found a significant, albeit low-sized, relation between the ε4 allele of apolipoprotein E and psychosis in Alzheimer’s disease [14].
As for serotonin receptors, the polymorphisms 102-T/C of serotonin receptor 2A (5-HTR2A) and Cys23Ser of serotonin receptor 2C (5-HTR2C) have been significantly associated with auditory and visual hallucinations in people with Alzheimer’s disease. The C102 alleles in the first polymorphism and Ser23 in the second lead to a lower expression of serotonin receptors that would only be associated with psychotic symptoms in a more advanced context of the disease. That is, once the neurodegeneration process is evident [25].
From an epigenetic perspective, Pishva et al. [26] conducted a case-control study in postmortem brain tissue, analyzing three regions associated with psychosis: superior temporal gyrus, entorhinal cortex, and prefrontal cortex. Using a complete epigenomic scanning association analysis in 29 brains of people with Alzheimer’s disease with psychosis and 18 without psychosis, they found differences in the methylome of different regions. They detected differences in the arsenic three methyltransferase gene (AS3MT) associated with neuronal development and differentiation. At the clinical level, some variants have been linked to the risk of schizophrenia.
Neurobiological evidence
Neurotransmitter systems
The cholinergic system presents interneurons that modulate the action of GABAergic and glutamatergic neurons in the hippocampus, prefrontal, parietal, and occipital cortex. The latter has an associative role in vision. The basal nucleus of Meynert would be atrophied in Parkinson’s disease carriers with visual hallucinations. Reduced levels of acetylcholine in cortical areas may cause information processed unconsciously to emerge consciously as hallucinations. In addition, anticholinergic drugs can cause visual hallucinations, while the opposite occurs with cholinesterase inhibitors, such as rivastigmine [27]. As for the serotonergic system, atrophy of some of its structures, such as the raphe nuclei, has been found in people with Parkinson’s disease and psychosis [27].
On the other hand, it has been reported that serotonergic 5-HT2A receptor agonists, such as lysergic acid diethylamide (LSD) or psilocybin, produce psychotic symptoms [28]. In contrast, pimavanserin, a 5-HT2A receptor inverse agonist or antagonist (without affinity for dopaminergic, histaminergic, muscarinic, or adrenergic receptors), exhibits specific antipsychotic effects in psychosis in dementia. By decreasing the activity of these receptors, regulating the excess cortical serotonergic activity, the GABAergic deficit, and decreasing glutamatergic activity towards the ventral tegmental area [29].
In the same line, people with Parkinson’s disease with visual hallucinations have increased 5-HT2A receptor expression in the ventral visual pathway, dorsolateral prefrontal cortex, orbitofrontal, and insula, a fact that could be related to psychotic symptoms. Indeed, it has been documented that excessive 5-HT2A receptor activity in the visual cortex can specifically lead to visual hallucinations. According to the dopaminergic hypothesis of psychosis, stimulation of the receptor in the prefrontal cortex activates N-methyl-D-aspartate (NMDA) receptors located on glutamatergic neurons, activating dopaminergic neurons in the ventral tegmental area and generating psychotic symptomatology [27]. The most altered areas in neurodegenerative processes coincide with areas with greater expression of this receptor (frontal, parietal, temporal, occipital lobes, and entorhinal cortex). Depending on which neurons are lost or damaged, this cortico-limbic pathway is altered through different mechanisms. This determines different phenomena that converge in common pathways, such as dysfunction of GABA interneurons, excessive serotonin availability, increased 5-HT2A receptor expression due to loss of serotonergic terminals in prefrontal and visual cortex, excess striatal dopaminergic activity, increased D2 receptor expression and glutamatergic hyperactivity, among others [29].
The dopaminergic hypothesis in schizophrenia suggests that delusions and hallucinations are caused by mesolimbic dopaminergic hyperactivity [30]. Although there is indirect evidence from functional neuroimaging studies, it is unclear whether this theory applies to the psychotic symptomatology observed in neurocognitive disorders. On the other hand, excess striatal dopamine at dopaminergic D2 and D3 receptors is associated with the development of delusions. Likewise, in samples of postmortem patients with Alzheimer’s disease, a higher density of D3 receptors in the nucleus accumbens and decreased dopaminergic neurotransmission in the amygdala have been verified [31]. Also, L-dopa, a dopaminergic agonist used in the treatment of Parkinson’s disease, can cause psychosis as an adverse effect [32].
In Alzheimer’s disease, evidence of dopaminergic degeneration has been found in the ventral tegmental area, even prior to the accumulation of β-amyloid peptide deposits and cell death in the hippocampus. Neurodegeneration is earlier in the ventral tegmental area than in other centers, such as the locus coeruleus, possibly because these neurons are more susceptible to cell death. Thus, degeneration in the mesolimbic or mesolimbic-cortical pathway is associated with neuropsychiatric symptoms in both Alzheimer’s disease and Parkinson’s disease [33]. Therefore, depending on the cause of neuronal degeneration, there may be dysfunction of GABAergic neurons, NMDA glutamate receptor, up-regulation of D2 receptor in the striatum or 5-HT2A receptor, as well as striatal denervation (causing hypersensitivity of mesolimbic and cortical dopaminergic receptors). Through any of these pathways, the generation of psychotic symptoms would be reached [28,32,34].
The serotonergic 5-HT2A receptor is present in both pyramidal and GABAergic cells. There is an excitatory/inhibitory balance depending on this receptor’s activation duration. In neurodegenerative processes, as the function of the GABAergic interneurons is lost, excessive excitation is triggered, which can lead to psychotic symptoms [29]. There would be an imbalance between cortical and mesolimbic excitatory tone, associated with hyperactivation of pyramidal neurons in the visual area and mesolimbic pathway, causing delusions and hallucinations [28]. In people with Alzheimer’s disease, amyloid plaques increase the sensitivity of glutamate receptors in GABAergic neurons, leading to overstimulation and GABAergic neuronal degeneration. In later stages, this phenomenon leads to glutamate NMDA receptor hypofunction. From pharmacological evidence, memantine, a drug used in the management of dementia, antagonizes the NMDA receptor, which aims to inhibit its excitotoxic effects. However, it may produce psychotic symptoms or worsen them [28].
Alterations in structural and functional neuroimaging studies
Psychotic symptoms in dementias have been associated with neurodegeneration in regions such as the dorsolateral frontal cortex, parietal, anterior cingulate, and ventral striatum. This may be linked to amyloid plaques, τ protein deposits, vascular alterations, chronic inflammation, or oxidative stress. In the case of the frontal lobe, its dysfunction causes alterations in perception, a fact that would contribute to the appearance of visual hallucinations [29]. Hyperactivity of the default mode network, mainly in frontoparietal regions, may lead to the appearance of false images originating from previously stored preconceptions [35].
In the case of Parkinson’s disease, a volumetric decrease of areas of the dorsal visual pathway involved in visual and cognitive processing has been found, as well as a reduction of the hippocampal volume, which is related to an alteration in the integration of information from ventral and dorsal visual networks. According to single-photon emission computed tomography (SPECT) studies, there would also be reduced connectivity between the hippocampus and occipitotemporal areas [35]. In a four-year cohort of patients with Parkinson’s disease, the subgroup with early-onset hallucinations (n = 21) had greater visual deficits and parietal, occipital, frontal, and hippocampal cortical atrophy [36].
As in Parkinson’s disease, patients with Lewy body disease have a higher density of Lewy bodies in areas of visual and auditory processing and integration, such as the parietal, frontal, and temporal cortex, as well as in the amygdala and the parahippocampal region. In the case of the parietal lobe, this is related to the perception and processing of external stimuli, contributing to the balance between two groups of attention-related neural networks: top-down and bottom-up processing networks [27]. A smaller gray matter volume has also been found in the occipital, occipitotemporal, medial, frontal, and inferior parietal areas. At the clinical level, more deficits have been found in executive functions, visual attention, and semantic memory [35]. According to positron emission tomography studies, there would be less metabolism in the precentral, superior, frontal, and parietal gyrus involving the ventral premotor cortex. Finally, reduced functional connectivity between bilateral cortical regions has been described [37].
Regarding Alzheimer’s disease with psychosis, a positron emission tomography study in 23 people with this pathology reported an increase in the number of striatal D2 and D3 receptors in those who had psychotic symptoms. Likewise, there would be a decrease in the ligand binding of the cholinergic muscarinic receptors M1, M3, M4, and M5. In addition, there would be decreased anticholinesterase activity in the frontotemporal area and increased M2 muscarinic receptor density in the orbitofrontal gyrus and temporal cortex [31].
Other findings include a higher density of neurofibrillary tangles of phosphorylated τ protein in the frontal cortex and cerebrospinal fluid [31] and lower brain flow in the right angular gyrus and right occipital lobe [38]. A cortico-subcortical circuit, including the frontal and cingulate gyrus, is associated with complex intentional and motivated behaviors and other higher-order functions such as planning and problem-solving. Disconnection between this and other neocortical circuits would be a factor related to psychosis in Alzheimer’s disease [38]. In the clinical spectrum comprising patients with criteria of frontotemporal dementia and amyotrophic lateral sclerosis who additionally present psychotic symptoms, a lower density of gray matter in the prefrontal cortex, bilateral superior temporal gyrus, and inferior frontal gyrus has been reported, compared to those without psychosis [39].
Finally, a systematic review conducted by Murdy-Rootes et al. [32] aimed to find clinical and structural differences in delusions in patients with bipolar affective disorder, schizophrenia, Alzheimer’s disease, Parkinson’s disease, and frontotemporal dementia, described that there would be common structural alterations. These include reductions in the gray matter of the dorsolateral prefrontal cortex, hippocampus, insula, superior temporal gyrus, and middle frontal gyrus.
Cognitive aspects of delusions in neurocognitive disorders
Delusions can occur in older persons without cognitive impairment and without other psychiatric diagnoses, which poses a nosological problem. It is not clear whether they could correspond to non-cognitive symptoms of neurodegenerative diseases, which manifest prior to cognitive alterations. It is also unclear whether they increase the risk of developing dementia [12].
Regarding the influence of cognitive impairment on delusions, the greater the cognitive deficit, the greater the risk of delusions, especially when these are related to frontal lobe dysfunction. However, a minimum of preserved cognitive function is required to elaborate delusions [40,41]. The type of cognitive impairment and environmental changes may influence the content of delusions. For example, memory problems or disorientation may influence the elaboration of delusions of theft or nonrecognition of home, and changes in the relationship with caregivers may promote paranoid thinking by not recognizing them. In this sense, some authors have considered delusions as part of the cognitive symptoms of dementia rather than an independent phenomenon [42]. Delusions in persons with severe cognitive deficits may overlap clinically with confabulations. In all false narratives, there would be dysfunction of frontal limbic areas and difficulty in assessing the veracity of the information retrieved. When patients accept false narratives as accurate, this frontal dysfunction involves problems in self-monitoring mediated by medial and orbitofrontal frontal regions. According to source and temporal context monitoring, the ventromedial prefrontal cortex participates in the retrieval of the "correct" feeling narrative, ruling out an attribution to a false belief. In parallel, the orbitofrontal cortex would be in charge of suppressing the interference of irrelevant thoughts, anticipations, and remnants of memories. Dysfunction in both areas would be necessary for confabulations to exist [43]. On the other hand, there is increasing evidence of lateralization of delusions in older people, a fact that would be linked to their content. For example, people with left-hemisphere dysfunction would present delusions with persecutory content, while right-hemisphere dysfunction would present mostly delusions of false recognition [40].
The two-factor theory attempts to explain the presence of delusions in neurocognitive disorders. This theory points out that a neuropsychological alteration initially promotes delirium (factor 1), and a second one interferes with the process of evaluation of the idea (factor 2). Without the latter disturbance, the delusional idea would be rejected. Factor 1 would be variable depending on the delirium, whereas factor 2 would be common to all delusions, and its biological basis would be the dysfunction of the right dorsolateral prefrontal cortex. In evaluating a hypothesis, tests are performed for formulation, confirmation, and disconfirmation. According to functional magnetic resonance studies, the dorsolateral prefrontal cortex is activated bilaterally at the beginning of this process. However, if feedback contradicts the hypothesis, the right area would be mainly activated [44]. For example, in Capgras delirium, or the belief that an imposter has replaced a relative, an alteration in the autonomic response to familiar faces has been evidenced by damage to the ventromedial prefrontal cortex (factor 1). But this damage alone is not sufficient to cause delirium since it has to coexist with damage to the right dorsolateral prefrontal cortex (factor 2), causing a "disconnection" between the temporal lobe (the area that coordinates facial processing) and the limbic circuits associated with appropriate personal and emotional stimuli [44,45]. In the case of Fregoli’s delirium, or the belief that there are familiar people following the person but they are "disguised", factor 1 could correspond to left temporo-occipital damage, causing hypersensitivity of the facial recognition system ("hyperfamiliarity" to unfamiliar faces). In contrast, factor 2 remains damage to the right dorsolateral prefrontal cortex [44,45,46]. Figure 1 synthesizes the main genetic, neurobiological, and cognitive findings related to psychosis in dementia.
Main genetic, neurobiological, and cognitive factors involved in the psychotic symptomatology of dementia.
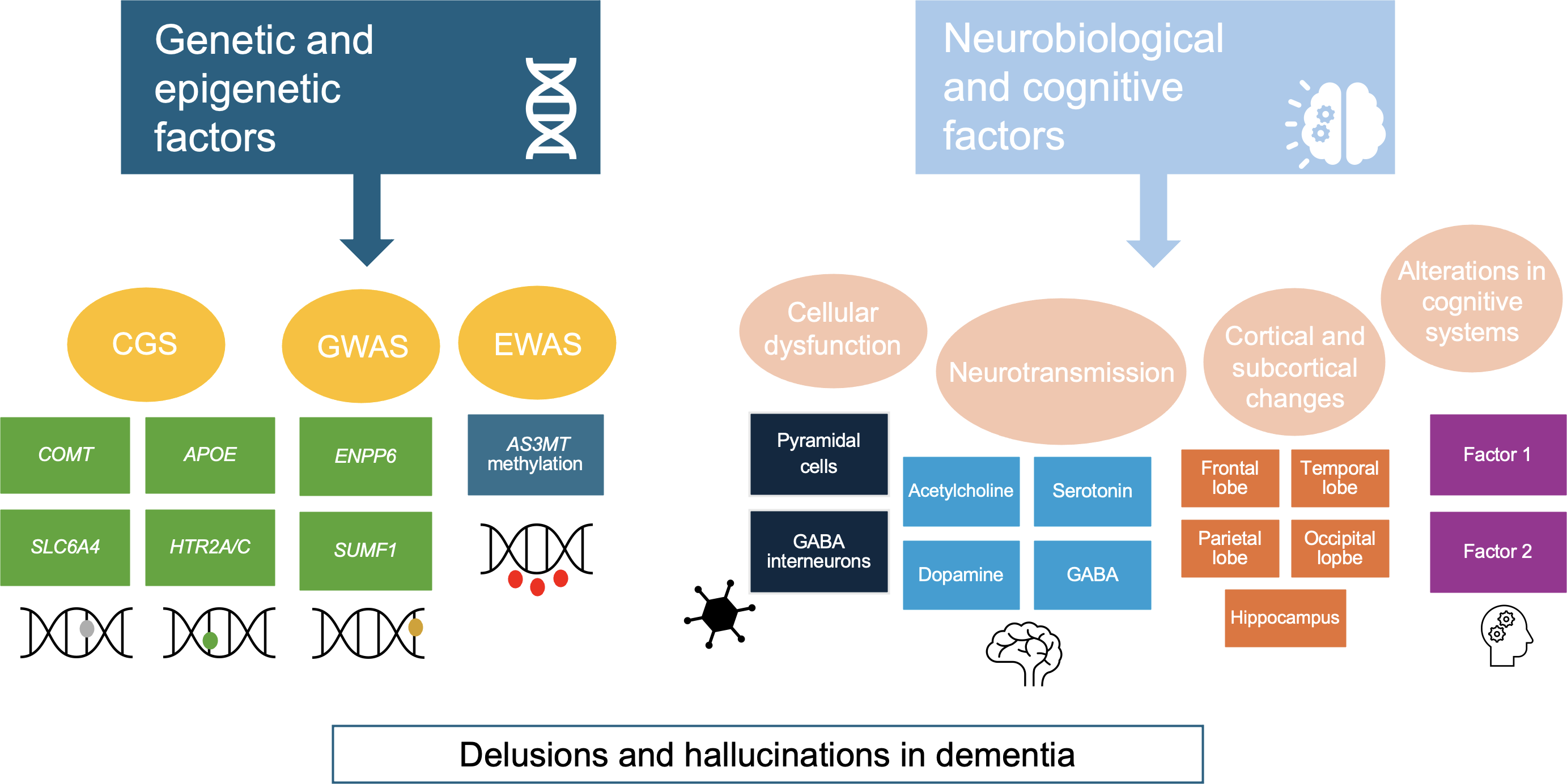
Source: Prepared by the authors based on the findings of the review.
Conclusions
Psychosis, whose cardinal symptoms are delusions and hallucinations, is a phenomenon that is mainly described in schizophrenia, although it is frequently observed in dementias. Most of the studies on psychosis in neurocognitive disorders have been conducted in samples of people with Alzheimer’s disease. Although classically, it has been considered that psychotic symptomatology appears in advanced stages of dementia, evidence has shown that it may even precede the onset of cognitive symptomatology. In this sense, the relationship between cognitive and psychotic symptomatology is complex and intertwined.
Multiple factors are involved in the genesis of psychotic symptoms during neurocognitive disorders, including genetic risk variants, dysfunction of neurotransmitter systems resulting from the neurodegenerative process, and brain morphofunctional changes, especially prefrontal, frontostriatal, and limbic.
Elucidating the biological basis of psychosis will allow changes in the clinical description and semiology of dementia, eventually providing a more timely diagnosis, hand in hand with specialized and person-centered therapeutic interventions. This is because although multiple biological alterations may converge in the same clinical symptomatology, those biological alterations may require different interventions. More research is warranted to completely understand these aspects that appear fundamental in pathology, which profoundly impacts patients' quality of life and environment, whose treatment is minimal.
