Artículo de revisión
← vista completaPublicado el 1 de febrero de 2012 | http://doi.org/10.5867/medwave.2012.02.5285
Tópicos sobre displasias óseas
Topics about bone dysplasia
Resumen
Es importante saber reconocer a las displasias esqueléticas como un grupo heterogéneo de patologías, cuya clasificación es bastante extensa y presenta limitaciones. Por lo anterior, es muy importante obtener mediciones antropométricas y un estudio esquelético completo para poder delinear adecuadamente el fenotipo, y así identificar el grupo diagnóstico al cual pertenece cada paciente y, en lo posible, establecer el diagnóstico. El diagnóstico de los casos que no pertenecen a las patologías más conocidas y comunes, como el grupo de la acondroplasia, presenta desafíos mayores y requiere de un enfrentamiento diagnóstico multidisciplinario.
Introducción
La talla baja es un motivo frecuente de consulta en pediatría. Se habla de talla baja cuando la relación talla/edad (T/E) es menor a 2 desviaciones estándar (DS) de lo esperado para su género, mientras que se clasifica de talla baja severa cuando la relación T/E es menor a 3 DS de lo esperado para su género. Las causas de talla baja son variadas, incluyendo casos de talla baja familiar, retraso de maduración ósea constitucional, trastornos endocrinológicos, genéticos y otras enfermedades crónicas. Las displasias óseas son un grupo de trastornos de origen genético en esencia monogénicos, que se deben sospechar, en especial en la presencia de desproporción de los segmentos corporales.
¿Cómo se define la displasia ósea o displasia esquelética?
Existen varias definiciones, pero la más completa es la que define la displasia ósea como una anomalía primaria o intrínseca en el desarrollo, crecimiento y mantenimiento del esqueleto humano. El término displasia esquelética no es un diagnóstico per se, sino que agrupa diferentes entidades que suelen presentar superposición entre sí.
La prevalencia general del grupo de las displasias esqueléticas se describe entre 1,1 a 7,6 por 10.000 nacimientos, dependiendo de la serie y del estudio. En término medio, se hace referencia a 2 por 10.000 nacimientos.
Antecedentes en la historia
Al menos desde Homero, en la Ilíada, se vienen describiendo casos de talla baja severa, que en ese tiempo se les llamaba a todos genéricamente como pigmeos. Están también representados en algunas vasijas de la cultura egipcia y de griega (Figura 1), así como varios siglos después, en grabados de algunas comunidades germánicas y nórdicas
 Tamaño completo
Tamaño completo Varios siglos después, artistas como Velásquez en “Las Meninas” y en varias obras más, representaron a personas con talla baja severa, que a menudo eran adoptadas por las cortes europeas (Figura 2)
 Tamaño completo
Tamaño completo Figura 2. Diferentes personajes de las cortes europeas, representados por Diego de Velásquez en sus obras:
a) “El bufón don Sebastián de Morra”
b) “Don Antonio, el inglés”
c) “El niño de Vallecas”
d) “El príncipe Baltasar Carlos y un enano”.
Por otro lado, algunos artistas también tenían talla baja y fueron retratados, como Toulouse-Lautrec (Figura 3), que se piensa tenía una displasia esquelética llamada Picnodisostosis.
 Tamaño completo
Tamaño completo Antecedentes desde los aspectos clínicos
Durante mucho tiempo se clasificaba a los individuos de talla baja solamente en base a si esta era o no proporcionada. En el caso de existir desproporción, si esta era de predominio de extremidades o del tronco. De esa manera las categorías comúnmente utilizadas hasta hace 50 años atrás eran:
- “Enanismo Hipofisiario”: talla baja pero proporcionada
- “Acondroplasia”: talla baja desproporcionada con menor crecimiento relativo de extremidades
- “Enfermedad de Morquio”: talla baja desproporcionada con menor tamaño relativo del tronco.
En base a análisis antropométricos, radiológicos e histopatológicos, que siguen siendo de utilidad para un enfoque diagnóstico inicial, se empezó a identificar que en realidad esto era una sobre simplificación, ya que coexistían diversas entidades diferentes.
A continuación comentaremos algunos elementos clínico-radiológicos que son de utilidad en el enfoque inicial de este grupo de pacientes.
Enfoque inicial: diferenciación entre talla baja normal o patológica
En un primer paso se debe distinguir entre una variante normal o patológica, para lo cual es importante disponer de la talla de ambos progenitores y así calcular la talla blanco para la adultez. De esa manera se puede evaluar si la curva de crecimiento está dentro de lo esperado según la maduración ósea del paciente. Asimismo se debe descartar algunas enfermedad sistémica asociada (endocrina, cromosómica, monogénica, teratogénica, otras).
Si se detecta una desproporción de los miembros y del tronco, hay que considerar la posibilidad de una displasia esquelética. Sin embargo, en las displasias óseas no siempre se observa talla baja desproporcionada, se pueden manifestar como talla baja proporcionada en rango leve, talla normal o incluso algunas entidades presentan talla alta o sobrecrecimiento.
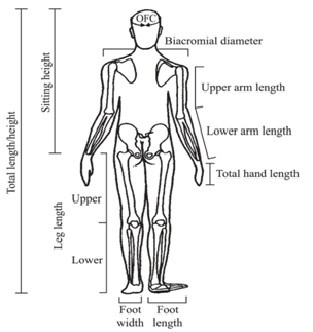
Para objetivar, es de relevancia realizar mediciones seriadas de la talla, perímetro cefálico, además considerar mediciones adicionales (Figura 4) para los cuales existen tablas y curvas según edad (ver referencias).
 Tamaño completo
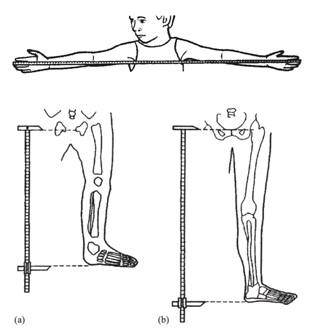
Tamaño completo Una medición de utilidad es la envergadura, que se obtiene de la punta entre ambos dedos con el paciente dispuesto contra una pared o una superficie sólida. En el caso de niños pequeños se les puede colocar en posición prona para que estén con las extremidades bien extendidas. Además, se recomienda medir el segmento inferior (SI), desde la sínfisis pubiana hasta el suelo o la planta de los pies, y el segmento superior (SS) se calcula por la diferencia de la talla (Figura 5). Las relaciones entre talla/envergadura y SS/SI según la edad, permiten objetivar si hay desproporción de los segmentos corporales.
 Tamaño completo
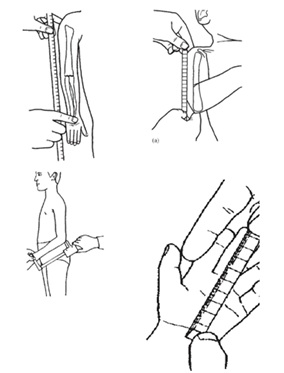
Tamaño completo Cuando el predominio es de extremidades, es útil determinar si esta se debe principalmente a la disminución del segmento proximal de las extremidades (húmero y fémures) o rizomélica; si es predominante de los segmentos medios de las extremidades (antebrazo o piernas propiamente tales) o mesomélica; o si es predominantemente de los segmentos más distales (manos y pies) o acromélica. Para objetivar esta impresión clínica existen puntos de referencia y sus respectivas curvas o tablas según género y edad (Figuras 6 a 9).
 Tamaño completo
Tamaño completo 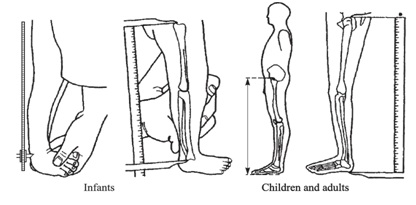 Tamaño completo
Tamaño completo 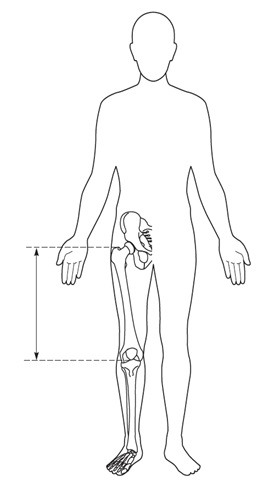 Tamaño completo
Tamaño completo 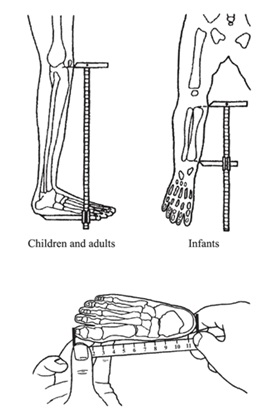 Tamaño completo
Tamaño completo Nomenclatura radiológica
En relación a las anomalías radiológicas una distinción básica se realiza entre las osteocondrodisplasias y las disostosis. Las osteocondrodisplasias se refieren a alteraciones generalizadas a nivel óseo y del cartílago, mientras que la disostosis se refiere a anomalías en huesos específicos o grupos de huesos relacionados, no afectando el esqueleto en su totalidad.
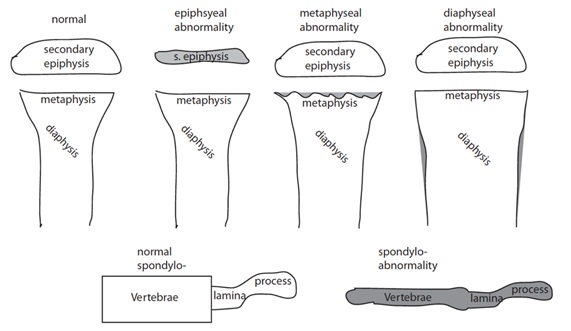
Dentro del análisis radiológico se debe identificar si las anomalías comprometen a las metáfisis, epífisis, diáfisis y/o las vértebras (espóndilo) (Figura 10).
 Tamaño completo
Tamaño completo Estudio esquelético de las displasias óseas
La recomendación más generalizada es llevar a cabo un estudio completo del esqueleto.
- Cráneo. Dos proyecciones: AP y lateral. Algunos promueven también una proyección para ver el foramen.
- Columna cervical, o columna total.
- Tórax, idealmente con una proyección costal.
- Pelvis.
- Extremidades superiores e inferiores. Algunos promueven que sea unilateral solamente, pero que si existen asimetrías, sea bilateral.
- Manos.
- Pies. Menos recomendado porque es muy difícil encontrar anomalías que no se vean en las manos.
Los controles radiológicos no se deben realizar muy seguido, sino dejando un intervalo de por los menos 12 meses. La Tabla I muestra las recomendaciones de estudio radiológico según los diferentes autores.
| Kant (2007) | ISDR (2004) | ESDN (2003) | Offiah (2003) | ACR (2001) | Mortier (2001) | Lachman(1998) | ||
| Cráneo | AP | + | + | - | + | + | + | +, Towne |
| L | + | + | - | + | + | + | + | |
| Columna cervical | AP | + | - | + | + | + | + | + |
| L | + | + | + | + | + | + | + | |
| Columna dorsolumbar | AP | + | + | + | + | + | + | + |
| L | + | + | + | + | + | + | + | |
| Tórax | AP | + | + | + | + | +, L | + | + |
| Pelvis | AP | + | + | + | + | + | + | + |
| EESS | AP | + | + | - | + | +, bi | + | + |
| Manos | AP | + | +, bi | + | + | +, bi | + | +, bi |
| EEII | AP | + | + | rodilla (AP-L), bi | + | +, bi | +, rodilla (L) | + |
| Pies | AP | - | +, bi | - | - | +, bi | - | +, bi |
Tabla I. Recomendaciones de estudio radiológico según diferentes autores. ACR= American College of Radiology; AP= Antero Posterior; bi= bilateral; ESDN= European Skeletal Dysplasia Network; ISDR= International Skeletal Dysplasia Registry; L: Lateral (Basado en Kant SG, et al. Horm Res. 2007).
Nosología y clasificación de las displasias óseas
En las últimas décadas se han descubierto la causa de muchas de estas patologías y se han incorporado elementos moleculares y etiopatogénicos en la clasificación, sin embargo, la clasificación todavía tiene algunas limitaciones.
La clasificación fue revisada por última vez en 2010. Se incluyen anomalías que no son solo displasias esqueléticas, sino que además trastornos que tienen algún componente relevante en el esqueleto. Se describen 456 entidades, reunidas en 40 grupos, y de los cuales en 316 de estas entidades se ha encontrado una mutación en uno o más de 226 genes.
La Tabla II muestra un resumen de todos los grupos. Para el interés de esta revisión, nos enfocaremos solamente en el primero y más conocido: el grupo de la acondroplasia. Pero no es el único, incluso hay muchos, como los trastornos de depósito lisosomal.
GRUPOS | ||
| 1. FGFR3 (acondroplasia) | 21. Condro dysplasia punctata (CDP) | |
| 2. Colágeno tipo 2 y similares | 22. Displasias osteoscleróticas neonatales | |
| 3. Colágeno tipo 11 | 23. Densidad ósea aumentada (sin modificación de la configuración ósea) | |
| 4. Trastornos de sulfatación | 24. Densidad ósea aumentada con compromiso metafisiario y/o diafisiario | |
| 5. Perlecan | 25. Osteogénesis imperfecta y densidad ósea disminuida | |
| 6. Agrecan | 26. Mineralización defectuosa | |
| 7. Filamina y relacionados | 27. Enf. de depósito lisosomal con compromiso esquelético (disostosis múltiple) | |
| 8. TPRV4 | 28. Osteolisis | |
| 9. Displasias con costillas cortas (con o sin polidactilia) | 29. Desarrollo esquelético desorganizado | |
| 10. Displasias epifisiaria múltiple y pseudoacondroplasia | 30. Síndromes con sobrecrecimiento y compromiso esquelético | |
| 11. Displasias metafisiarias | 31. Osteoartropatías genéticas inflamatorias/similares a artritis reumatoide | |
| 12. Displasias espondilo metafisiarias (SMD) | 32. Displasia cleidocraneal y defectos de osificación aislados | |
| 13. Displasias espondilo-epi-(meta-)fisiarias [SE(M)D] | 33. Síndromes de craneosinostosis | |
| 14. Displasias espondilo displásicas severas | 34. Disostosis con compromiso craneofacial predominante | |
| 15. Displasias acromélicas | 35. Disostosis con compromiso vertebral y costal predominante | |
| 16. Displasia acromesomélica | 36. Disostosis rotulianas | |
| 17. Displasias mesomélicas y rizo-mesomélicas | 37. Braquidactilias (con o sin manifestaciones extra-esqueléticas) | |
| 18. Displasias con “huesos curvos” (bent bones) | 38. Defectos de reducción-hipoplasia de extremidades | |
| 19. Displasias con “huesos delgados” (slender bones) | 39. Polidactilia-Sindactilia-Trifalangismo | |
| 20. Displasias con luxaciones articulares múltiples | 40. Defectos en la formación articular y sinostosis | |
Tabla II. Nosología y clasificación de los trastornos esqueléticos de origen genético, revisión 2010 (Warman ML, et al. Am J Med Genet Part A. 2011).
Grupo 1: FGFR3
Este grupo toma el nombre del gen para el receptor III del factor de crecimiento de fibroblastos. Las mutaciones en este gen alteran la osificación endocondral (principalmente en la metáfisis de los huesos largos), generando un espectro de anomalías esqueléticas de severidad muy variable, con un patrón de herencia autosómico dominante. El fenotipo más reconocido es la acondroplasia, sin embargo, algunas mutaciones se pueden manifestar como individuos que pueden tener una talla que puede ser talla baja leve con leve acortamiento de las extremidades y macrocefalia (hipocondroplasia). Hacia el otro extremo, hay casos más severos que son letales por hipoplasia pulmonar (Displasia tanatofórica). También se puede dar una patología de severidad intermedia con acortamiento de predominio rizomélico yacantosis nigricans (SADDAN).
Gen FGFR3
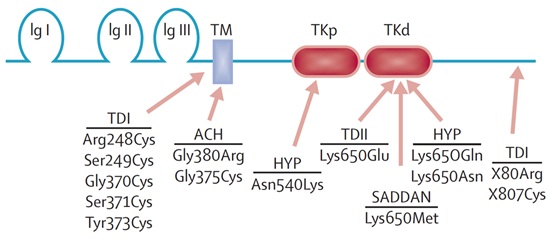
La Figura 11 muestra la estructura del gen, con dominios similares a inmunoglobulina (Ig), el dominio transmembrana (TM), y los dominios intra-citoplasmáticos tirosina kinasa (TK). Mutaciones a lo largo de todo el gen pueden dar los fenotipos anteriormente descritos. La particularidad de la acondroplasia es que prácticamente todos los pacientes tienen mutaciones en el mismo dominio, que es el transmembrana, y sobre el 95% tienen exactamente la misma mutación (Gly380Arg).
 Tamaño completo
Tamaño completo La Figura 11 muestra un esquema de la proteína codificada por el gen FGFR3. Las flechas indican el lugar donde preferentemente ocurren las mutaciones asociados a cada uno de los fenotipos junto con el nombre de cada mutación. ACH: Acondroplasia; HYP: Hipocondroplasia; Ig I: Dominio similar a inmunoglobulina 1; Ig II: Dominio similar a inmunoglobulina 2; Ig III: Dominio similar a inmunoglobulina 3; TD I: Displasia tanatofórica tipo 1; TD II: Displasia tanatofórica tipo 2; TKd: Dominio tirosina kinasa distal; TKp: Dominio tirosina kinasa proximal; TM: Dominio transmembrana.
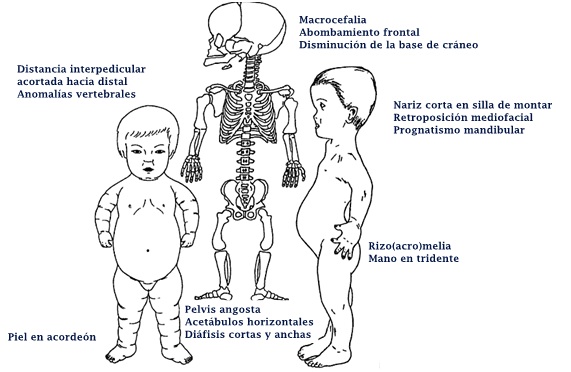
¿Qué es la acondroplasia?
La acondroplasia es una condición con talla baja severa, predominio de acortamiento rizomélico de extremidades, pero también hay acortamiento acromélico (Figura 12). Debido a este acortamiento, la piel se describe como un acordeón por el exceso de pliegues (Figura 12). En las manos se describe una mano en tridente, donde la separación entre el segundo, tercer y cuarto dedo es similar (Figura 14a), no es asimétrica como en las manos no afectadas por estas alteraciones. Se acompaña de macrocefalia, con abombamiento frontal y un pequeño foramen magno. También se caracteriza por una nariz corta, en silla de montar y retroposición de las estructuras medias de la cara y prognatismo mandibular (Figura 12). Suele haber hiperlaxitud articular generalizada. Esto se puede evaluar en los lactantes en posición sentada se puede tomar y juntar las plantas al mismo nivel de las caderas y eso se ve en más del 90% de los niños con acondroplasia. Este signo se llama “de las plantas” o de Scott (Figura 13).
A nivel radiológico, se busca dirigidamente en la toma antero posterior de la columna, la distancia existente entre los pedículos vertebrales de proximal hacia distal. Normalmente esto tiende a ensancharse, pero en el caso de la acondroplasia y la hipocondroplasia, esto se mantiene o decrece en longitud (Figura 14b). En la visión lateral de columna suele haber exageración de la lordosis lumbar al momento de comenzar a caminar. En niños mayores puede desarrollarse cifosis toracolumbar, lo que se puede asociar a anomalías vertebrales congénitas (Figura 14c). En la pelvis se puede apreciar una pelvis angosta, cuadrada, con los acetábulos horizontales (Figura 14b), mientras que en los hombros puede haber incompleta formación de la fosa glenoidea escapular. En los huesos largos se pueden ver las diáfisis cortas y anchas con ensanchamiento metafisiario.
 Tamaño completo
Tamaño completo 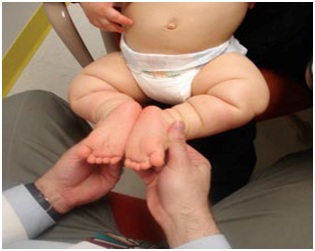 Tamaño completo
Tamaño completo 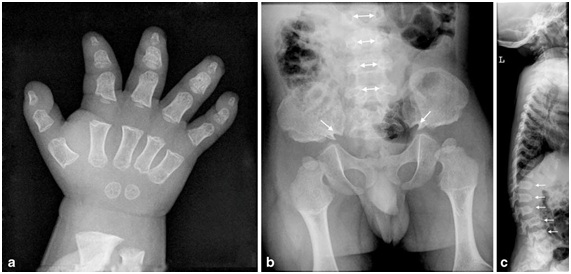 Tamaño completo
Tamaño completo En la Figura 14 se muestra: a) Mano en tridente, huesos tubulares cortos; b) Disminución de la distancia interpedicular hacia distal (flechas con doble cabeza); Pelvis con alas ilíacas cuadradas, techos acetabulares horizontales y escotadura ciática angostas bilaterales (flechas de una cabeza); c) Vertebrales con forma de bala (flechas)
Notas
Se agradece al Hospital Padre Hurtado de la ciudad de Santiago, Chile, por la colaboración brindada para la realización de este artículo.
