Reporte de caso
← vista completaPublicado el 5 de abril de 2017 | http://doi.org/10.5867/medwave.2017.03.6901
Dermatosis inmunoglobulina A lineal: a propósito de un caso
Linear immunoglobulin A dermatosis: A case report
Resumen
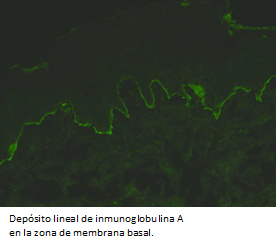
En este texto se presenta el caso de una paciente de sesenta y cinco años con sintomatología de dos meses de evolución consistente en prurito y lesiones hiperpigmentadas anulares en tronco, glúteos y extremidades superiores. En el área flexora de las muñecas presenta vesículas sobre base eritematosa y piel sana, sin evidencia de compromiso mucoso. Al estudio histológico se constata dermatitis vesicular subepidérmica con infiltrado inflamatorio de neutrófilos y eosinófilos. La inmunofluorescencia directa muestra depósito lineal y continuo de inmunoglobulina A en zona de membrana basal, compatible con dermatosis por inmunoglobulina A lineal.
Introducción
La dermatosis por inmunoglobulina A lineal es un trastorno autoinmunitario poco frecuente, que se caracteriza por la presencia de lesiones vesiculosas y ampollares. Su diagnóstico se confirma con la realización de inmunofluorescencia directa, que muestra la presencia de depósitos de inmunoglobulina A en la unión dermoepidérmica [1]. Su incidencia en Sudamérica aún se desconoce.
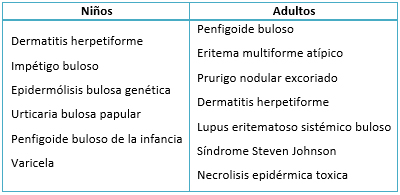
A continuación exponemos el presente caso clínico, debido a la baja incidencia de esta patología en nuestra población y a su amplio diagnóstico diferencial, tanto en población pediátrica como adulta. Además, presenta una conducta terapéutica específica en comparación a otras dermatosis ampollares autoinmunes.
Presentación de caso clínico
Paciente femenino de 65 años de edad, con antecedentes de adenoma suprarrenal, hipertensión arterial y enfermedad pulmonar obstructiva crónica secundaria a tabaquismo en tratamiento con atenolol. Refiere cuadro de dos meses de evolución, caracterizado inicialmente por prurito intenso y posterior aparición de lesiones eritematosas asociadas a vesículas, en tronco y extremidades superiores. Éstas aumentaron de tamaño en forma progresiva, adquiriendo algunas una distribución anular y que posteriormente evolucionaron a costras periféricas.
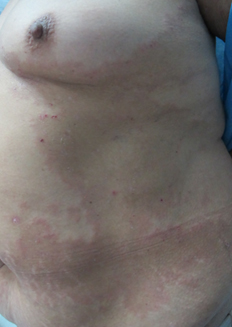
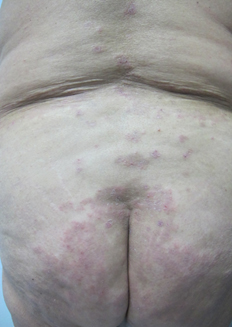
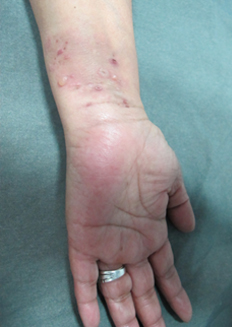
Al examen físico destacó extensa placa hiperpigmentada anular en tronco (Figura 1), de forma irregular, con borde eritematoso y erosiones costrosas de predominio periférico. Presentó lesiones de características similares de menor tamaño en dorso, glúteos (Figura 2) y extremidades superiores. En superficies flexoras de ambas muñecas se asoció a ampollas y vesículas tensas, de contenido seroso, sobre base eritematosa y piel sana (Figura 3). No se observó compromiso de mucosas.
Debido a estos antecedentes clínicos, se planteó como hipótesis diagnóstica dermatosis inmunoglobulina A lineal, por lo que se planificó biopsia incisional para su confirmación. Se comenzó a suministrar 30 milígramos de prednisona por vía oral, después de la obtención de la muestra.
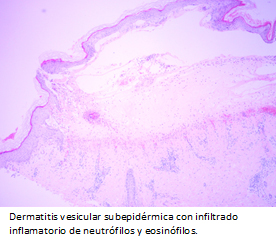
El estudio histológico con tinción de hematoxilina-eosina, evidenció dermatitis vesicular subepidérmica con infiltrado inflamatorio de neutrófilos y eosinófilos (Figura 4), concordante con enfermedad ampollar autoinmune subepidérmica. La inmunofluorescencia directa mostró depósito lineal y continuo de inmunoglobulina A en zona de membrana basal (Figura 5), compatible con dermatosis por inmunoglobulina A lineal.
Examenes de control en rangos de normalidad y actividad de glucosa 6 fosfato deshidrogenasa normal. Se inició tratamiento con 50 miligramos y 100 miligramos de dapsona día por medio, evolucionando con hiperpigmentación post inflamatoria asimétrica en dorso, flanco y axila derecha, sin nuevas lesiones ampollares. Por este motivo, se redujo progresivamente la dosis de prednisona oral hasta la suspensión y se disminuyó dosis de dapsona a 50mg al día, con respuesta clínica favorable.

Autores
Fernando Valenzuela Ahumada
Facultad de Medicina, Universidad de Chile, Santiago, Chile
Departamento de Dermatología, Hospital Clínico Universidad de Chile, Santos Dumont 999, Independencia, Santiago, Chile
Roberto Bustos Macaya
Facultad de Medicina, Universidad de Chile, Santiago, Chile
Gabriela Paz Romero Morgado
Facultad de Medicina, Universidad de Chile, Santiago, Chile
Margarita Sánchez Chacón
Facultad de Medicina, Universidad de Chile, Santiago, Chile
Figuras
Foro
No hay mensajes
Historial
Citación Valenzuela Ahumada F, Bustos Macaya R, Romero Morgado GP, Sánchez Chacón M. Linear immunoglobulin A dermatosis: A case report. Medwave 2017;17(03):e6901 doi: 10.5867/medwave.2017.03.6901
Envío 08/11/2016
Aceptación 26/01/2017
Publicación 05/04/2017
Métricas del artículo
Artículo no tiene métricas.
Notas
Palabras claves
linear IgA bullous dermatosis, bullous disease
Origen y arbitraje
no solicitado. con revisión por tres pares revisores externos, a doble ciego
Referencias
- Venning VA. Linear IgA disease: clinical presentation, diagnosis, and pathogenesis. Immunol Allergy Clin North Am. 2012 May;32(2):245-53, vi. | CrossRef | PubMed
- Fortuna G, Marinkovich MP. Linear immunoglobulin A bullous dermatosis. Clin Dermatol. 2012 Jan-Feb;30(1):38-50. | CrossRef | PubMed
- Sandoval M, Farias MM, Gonzalez S. Linear IgA bullous dermatosis: report of five cases in Chile. Int J Dermatol. 2012 Nov;51(11):1303-6. | CrossRef | PubMed
- Zone JJ, Taylor TB, Kadunce DP, Meyer LJ. Identification of the cutaneous basement membrane zone antigen and isolation of antibody in linear immunoglobulin A bullous dermatosis. J Clin Invest. 1990 Mar;85(3):812-20. | PubMed
- Vodegel RM, de Jong MC, Pas HH, Jonkman MF. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol. 2002 Dec;47(6):919-25. | PubMed
- Conleth A, Zone J. Linear IgA bullous dermatosis. Int J Dermatol. 1999 Nov;38(11):818-27. | PubMed
- Fuentelsaz V, Campos M. Dermatosis IgA lineal de la infancia. Rev Pediatr Aten Primaria. 2013;15:141-5. | Link
- Chen S, Mattei P, Fischer M, Gay JD, Milner SM, Price LA. Linear IgA bullous dermatosis. Eplasty. 2013 Jul 2;13:ic49. | PubMed
- Ingen-Housz-Oro S. [Linear IgA bullous dermatosis: a review]. Ann Dermatol Venereol. 2011 Mar;138(3):214-20. | CrossRef | PubMed
- Collier PM, Wojnarowska F, Welsh K, McGuire W, Black MM. Adult linear IgA disease and chronic bullous disease of childhood: the association with human lymphocyte antigens Cw7, B8, DR3 and tumour necrosis factor influences disease expression. Br J Dermatol. 1999 Nov;141(5):867-75. | PubMed
- Patsatsi A. Chronic Bullous Disease or Linear IgA Dermatosis of Childhood –Revisited. J Genet Syndr Gene Ther. 2013;4:6 | CrossRef
- Hirako Y, Usukura J, Uematsu J, Hashimoto T, Kitajima Y, Owaribe K. Cleavage of BP180, a 180-kDa bullous pemphigoid antigen, yields a 120-kDa collagenous extracellular polypeptide. J Biol Chem. 1998 Apr 17;273(16):9711-7. | PubMed
- Zone JJ, Taylor TB, Meyer LJ, Petersen MJ. The 97 kDa linear IgA bullous disease antigen is identical to a portion of the extracellular domain of the 180 kDa bullous pemphigoid antigen, BPAg2. J Invest Dermatol. 1998 Mar;110(3):207-10. | PubMed
- Allen J, Wojnarowska F. Linear IgA disease: the IgA and IgG response to dermal antigens demonstrates a chiefly IgA response to LAD285 and a dermal 180-kDa protein. Br J Dermatol. 2003 Nov;149(5):1055-8. | PubMed
- Hendrix JD, Mangum KL, Zone JJ, Gammon WR. Cutaneous IgA deposits in bullous diseases function as ligands to mediate adherence of activated neutrophils. J Invest Dermatol. 1990 May;94(5):667-72. | PubMed
- Zone JJ, Egan CA, Taylor TB, Meyer LJ. IgA autoimmune disorders: development of a passive transfer mouse model. J Investig Dermatol Symp Proc. 2004 Jan;9(1):47-51. | PubMed
- Caproni M, Rolfo S, Bernacchi E, Bianchi B, Brazzini B, Fabbri P. The role of lymphocytes, granulocytes, mast cells and their related cytokines in lesional skin of linear IgA bullous dermatosis. Br J Dermatol. 1999 Jun;140(6):1072-8. | PubMed
- Mintz EM, Morel KD. Clinical features, diagnosis, and pathogenesis of chronic bullous disease of childhood. Dermatol Clin. 2011 Jul;29(3):459-62, ix. | CrossRef | PubMed
- Weinberg MA, Insler MS, Campen RB. Mucocutaneous features of autoimmune blistering diseases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997 Nov;84(5):517-34. | PubMed
- Shimizu S, Natsuga K, Shinkuma S, Yasui C, Tsuchiya K, Shimizu H. Localized linear IgA/IgG bullous dermatosis. Acta Derm Venereol. 2010 Nov;90(6):621-4. | CrossRef | PubMed
- Burge S, Wojnarowska F, Marsden A. Chronic bullous dermatosis of childhood persisting into adulthood. Pediatr Dermatol. 1988 Nov;5(4):246-9. | PubMed
- Verma R, Vasudevan B, Pragasam V, Dabbas D. Linear IgA disease in an adult with unusual clinical features. Indian Dermatol Online J. 2013 Apr;4(2):115-8. | CrossRef | PubMed
- Aultbrinker EA, Starr MB, Donnenfeld ED. Linear IgA disease. The ocular manifestations. Ophthalmology. 1988 Mar;95(3):340-3. | PubMed
- Godfrey K, Wojnarowska F, Leonard J. Linear IgA disease of adults: association with lymphoproliferative malignancy and possible role of other triggering factors. Br J Dermatol. 1990 Oct;123(4):447-52. | PubMed
- Girão L, Fiadeiro T, Rodrigues JC. Burn-induced linear IgA dermatosis. J Eur Acad Dermatol Venereol. 2000 Nov;14(6):507-10. | PubMed
- Vargas TJ, Fialho M, Santos LT, Rodrigues PA, Vargas AL, Sousa MA. Linear IgA dermatosis associated with ulcerative colitis: complete and sustained remission after total colectomy. An Bras Dermatol. 2013 Jul-Aug;88(4):600-3. | CrossRef | PubMed
- Collier PM, Kelly SE, Wojnarowska F. Linear IgA disease and pregnancy. J Am Acad Dermatol. 1994 Mar;30(3):407-11. | PubMed
- Camilleri M, Pace JL. Drug-induced linear immunoglobulin-A bullous dermatosis. Clin Dermatol. 1998 May-Jun;16(3):389-91. | PubMed
- Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001 Nov-Dec;19(6):719-27. | PubMed
- Reyes-Baraona F, Andino R, Carrasco JE, Arriagada C, Guerrero S. [Linear IgA bullous dermatosis of childhood: case report]. Arch Argent Pediatr. 2014 Apr;112(2):e57-60. | CrossRef | PubMed
- Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults. A comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol. 1988 Nov;19(5 Pt 1):792-805. | PubMed
- Willsteed E, Bhogal BS, Black MM, McKee P, Wojnarowska F. Use of 1M NaCl split skin in the indirect immunofluorescence of the linear IgA bullous dermatoses. J Cutan Pathol. 1990 Jun;17(3):144-8. | PubMed
- Kuechle MK, Stegemeir E, Maynard B, Gibson LE, Leiferman KM, Peters MS. Drug-induced linear IgA bullous dermatosis: report of six cases and review of the literature. J Am Acad Dermatol. 1994 Feb;30(2 Pt 1):187-92. | PubMed
- Bhogal B, Wojnarowska F, Marsden RA, Das A, Black MM, McKee PH. Linear IgA bullous dermatosis of adults and children: an immunoelectron microscopic study. Br J Dermatol. 1987 Sep;117(3):289-96. | PubMed
- Allen J, Wojnarowska F. Linear IgA disease: the IgA and IgG response to the epidermal antigens demonstrates that intermolecular epitope spreading is associated with IgA rather than IgG antibodies, and is more common in adults. Br J Dermatol. 2003 Nov;149(5):977-85. | PubMed
- Mintz EM, Morel KD. Treatment of chronic bullous disease of childhood. Dermatol Clin. 2011 Oct;29(4):699-700. | CrossRef | PubMed
- Wojnarowska F. Linear IgA dapsone responsive bullous dermatosis. J R Soc Med. 1980 May;73(5):371-3. | PubMed
- McFadden JP, Leonard JN, Powles AV, Rutman AJ, Fry L. Sulphamethoxypyridazine for dermatitis herpetiformis, linear IgA disease and cicatricial pemphigoid. Br J Dermatol. 1989 Dec;121(6):759-62. | PubMed
- Mervic L, Dragos V, Pavlović MD. Linear IgA bullous dermatosis of childhood: successful treatment with miocamycin and topical corticosteroid. Clin Exp Dermatol. 2009 Oct;34(7):e391-2. | CrossRef | PubMed
- Kasperkiewicz M, Schmidt E. Current treatment of autoimmune blistering diseases. Curr Drug Discov Technol. 2009 Dec;6(4):270-80. | PubMed
- Alajlan A, Al-Khawajah M, Al-Sheikh O, Al-Saif F, Al-Rasheed S, Al-Hoqail I, et al. Treatment of linear IgA bullous dermatosis of childhood with flucloxacillin. J Am Acad Dermatol. 2006 Apr;54(4):652-6. | PubMed
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993 Jun;28(6):998-1000. | PubMed
- Peoples D, Fivenson DP. Linear IgA bullous dermatosis: successful treatment with tetracycline and nicotinamide. J Am Acad Dermatol. 1992 Mar;26(3 Pt2):498-9. | PubMed
- Yomada M, Komai A, Hashimato T. Sublamina densa-type linear IgA bullous dermatosis successfully treated with oral tetracycline and niacianamide. Br J Dermatol. 1999 Sep;141(3):608-9. | PubMed
- Khan IU, Bhol KC, Ahmed AR. Linear IgA bullous dermatosis in a patient with chronic renal failure: response to intravenous immunoglobulin therapy. J Am Acad Dermatol. 1999 Mar;40(3):485-8. | PubMed
- Kroiss MM, Vogt T, Landthaler M, Stolz W. High-dose intravenous immune globulin is also effective in linear IgA disease. Br J Dermatol. 2000 Mar;142(3):582. Erratum in: Br J Dermatol 2000 Jun;142(6):1268. | PubMed
- Letko E, Bhol K, Foster CS, Ahmed AR. Linear IgA bullous disease limited to the eye: a diagnostic dilemma: response to intravenous immunoglobulin therapy. Ophthalmology. 2000 Aug;107(8):1524-8. | PubMed
- Lozinski A, Baum S, Sagi L, Volkov A, Trau H, Barzilai A. [Rituximab (Mabthera) for treatment of rare autoimmune bullous skin disorders]. Harefuah. 2012 Oct;151(10):562-5, 606. | PubMed
- Talhari C, Althaus C, Megahed M. Ocular linear IgA disease resulting in blindness. Arch Dermatol. 2006 Jun;142(6):786-7. | PubMed