Notas metodológicas
← vista completaPublicado el 8 de abril de 2020 | http://doi.org/10.5867/medwave.2020.02.7869
Conceptos generales en bioestadística y epidemiología clínica: estudios experimentales con diseño de ensayo clínico aleatorizado
General concepts in biostatistics and clinical epidemiology: Experimental studies with randomized clinical trial design
Resumen
En los estudios experimentales, el investigador aplica una intervención sobre los participantes, por lo que su eje temporal es prospectivo. Estas características permiten probar relaciones causales, pero implican una rigurosa evaluación bioética y una inscripción previa del protocolo de estudio. Entre las investigaciones experimentales se encuentran los estudios preclínicos y diversos ensayos con objetivos distintos según la fase de desarrollo en que se encuentre. Entre ellos se incluyen los ensayos clínicos, cuyo objetivo principal es evaluar la eficacia y la seguridad de alguna intervención o terapia. El diseño convencional de ensayo clínico es aleatorizado controlado enmascarado, donde se asigna aleatoriamente a los participantes a una intervención y a sus comparadores (otra intervención, placebo o nada); lo que permite controlar el sesgo de selección y el de confusión. Asimismo, los participantes y los investigadores no conocen la intervención que fue asignada. El análisis de sus resultados debe considerar a todos los sujetos originalmente aleatorizados, lo que se conoce como análisis por intención de tratar. Otros tipos de estudios experimentales incluyen los estudios cuasiexperimentales y los estudios experimentales con controles externos. Las medidas de magnitud del efecto utilizadas son el riesgo relativo, las reducciones absolutas y relativas de riesgo y los números necesarios a tratar y dañar. Si bien los factores de confusión son controlados por el proceso de aleatorización, otros sesgos que hay que considerar son los de selección, desarrollo, detección, desgaste y reporte. En este artículo, se abordan los conceptos teóricos generales sobre los estudios experimentales en seres humanos, fundamentalmente sobre los ensayos clínicos aleatorizados, considerando sus elementos fundamentales, desarrollo histórico, aspectos bioéticos, construcción, variantes en el diseño, medidas de asociación, sesgos y pautas de reporte. Finalmente, se sintetizan algunas consideraciones que siempre hay que tener en mente durante la ejecución y la evaluación de un ensayo clínico. Esta revisión es la quinta entrega de una serie metodológica sobre conceptos generales en bioestadística y epidemiologia clínica desarrollada por la Cátedra de Metodología de la Investigación Científica de la Escuela de Medicina de la Universidad de Valparaíso, Chile.
Ideas clave
- Los ensayos clínicos aleatorizados evalúan la eficacia y la seguridad de una intervención controlada sobre seres humanos, permitiendo establecer causalidad.
- Existen diversos tipos de ensayos clínicos aleatorizados según su finalidad.
- Controlan eficazmente el sesgo de confusión, pero pueden presentar otro tipo de sesgos.
- Existen pautas internacionales que regulan la conducción ética y el reporte de estos estudios.
Introducción
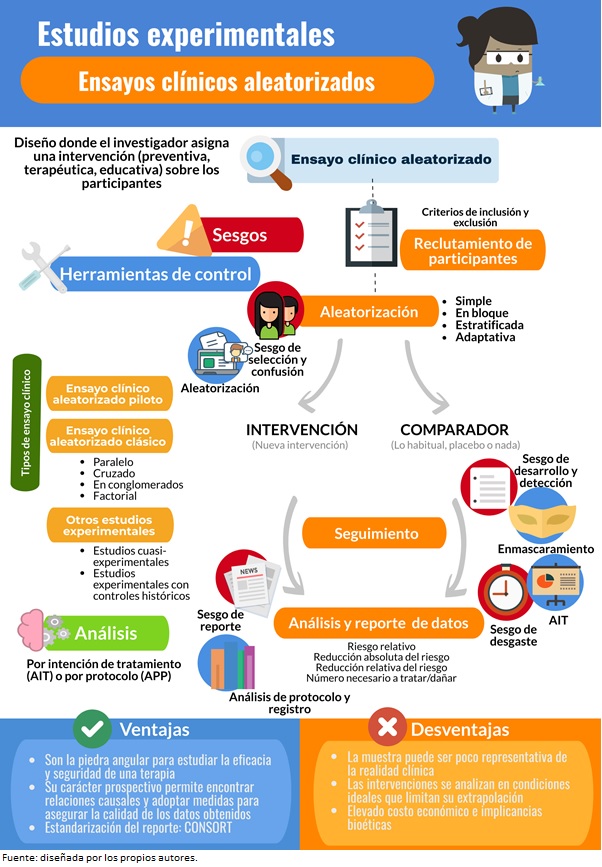
Los estudios experimentales son aquellos en que el investigador aplica una intervención sobre los participantes, como ha sido definido en un artículo anterior de esta serie metodológica[1]. Aquí se enmarcan todos los tipos de ensayo clínico que analizan intervenciones preventivas, terapéuticas, educativas, entre otras, que se realizan tanto sobre individuos como sobre grupos poblacionales[2],[3]. Algunos autores han considerado que las series de casos sin grupo control son el punto inicial de los estudios sobre intervenciones terapéuticas, ya que han aportado en el desarrollo de nuevas técnicas quirúrgicas y en el estudio de intervenciones en condiciones muy infrecuentes en donde la ejecución de un ensayo clínico se dificulta[4]. Sin embargo, los ensayos clínicos aleatorizados son el diseño metodológico de elección para evaluar la eficacia (el verdadero efecto biológico de una intervención) y la efectividad (el efecto de una intervención en la práctica clínica cotidiana)[5], además de ser el tipo de estudio primario con la mayor capacidad de controlar los sesgos[6].
En la década de 1920, Fisher conceptualizó la asignación aleatoria mediante la aleatorización de cultivos a recibir un tipo de semillas u otro. Más tarde, el Medical Research Council de Reino Unido adaptó la aleatorización a la epidemiología clínica, al realizar un ensayo clínico aleatorizado para evaluar el efecto de la estreptomicina entre portadores de neumonía, publicado en 1948[7], el que suele reconocerse como el primer ensayo clínico aleatorizado[8]. No obstante, ya en 1907 el médico William Fletcher había publicado los resultados de un ensayo clínico aleatorizado conducido para analizar el origen del beriberi en pacientes internados en un manicomio de Kuala Lumpur[9], donde las condiciones podían ser mejor controladas. Él asignó pacientes a alimentarse con arroz blanco o integral, de acuerdo con la teoría de la época que asociaba el beriberi al consumo de arroz blanco.
Durante los últimos 70 años, los ensayos clínicos se han perfeccionado, constituyéndose en la metodología fundamental de las agencias reguladoras de medicamentos para autorizar la comercialización de estos productos[10]. La investigación del potencial nocivo de algunos fármacos comienza a cobrar mayor relevancia a partir de problemáticas de salud pública: muerte súbita en pacientes anestesiados con cloroformo[11],[12], detección de aplasia de huesos largos en recién nacidos asociada al uso de talidomida en madres con náuseas y vómitos del embarazo[13] y, más recientemente, los efectos adversos del uso de ácido acetilsalicílico[2],[14]. Por lo tanto, diversas entidades sanitarias han decidido enfocar en la ejecución de ensayos clínicos gran parte del financiamiento disponible para la investigación en salud[10]. En el Ejemplo 1 se muestra un ensayo clínico aleatorizado.
Este artículo corresponde a la quinta entrega de una serie metodológica de seis revisiones narrativas acerca de tópicos generales en bioestadística y epidemiología clínica, las que exploran artículos publicados disponibles en las principales bases de datos y textos de consulta especializados. La serie está orientada a la formación de estudiantes de pre y posgrado. Es realizada por la Cátedra de Metodología de la Investigación Científica de la Escuela de Medicina de la Universidad de Valparaíso, Chile. El objetivo de este manuscrito es abordar los principales conceptos teóricos y prácticos de los estudios experimentales en seres humanos, fundamentalmente los ensayos clínicos aleatorizados.
Conceptos preliminares: control, aleatorización y cegamiento
Los ensayos clínicos corresponden a diseños experimentales prospectivos (se realiza un seguimiento en el tiempo), con la capacidad de establecer relaciones de causalidad, ya que corroboran que la causa (intervención) precede al efecto (desenlace), y que esta causa corresponde a una intervención administrada por los investigadores. El que un ensayo clínico sea “controlado”, implica que se presenta una comparación estadísticamente válida entre los resultados obtenidos en un grupo intervención y un grupo “control” o comparador. Si no se incluyera un grupo control, no podría asegurarse por qué el desenlace ocurrió[5]. Para esto, es necesario que se incluyan al menos dos grupos de pacientes y/o voluntarios sanos, cuya asignación a un tratamiento experimental o una intervención control será aleatoria; de ahí la calificación de ensayos clínicos “aleatorizados”. En el proceso de aleatorización no intervienen el investigador ni el sujeto en experimentación. La aleatorización es un fenómeno clave en este tipo de diseños, ya que es el mejor método de control de algunos de los sesgos asociados a la investigación con seres humanos. De hecho, la aleatorización ha sido considerada por algunos autores como el descubrimiento “más revolucionario y profundo de la medicina moderna”, ya que sobre ella descansan múltiples grandes descubrimientos: desde el uso de la penicilina hasta la terapia génica[8].
Entre los ensayos clínicos controlados se encuentran aquellos abiertos o “no ciegos”, donde el participante y el investigador conocen la intervención asignada (por ejemplo, en ensayos clínicos aleatorizados que analizan intervenciones quirúrgicas). Esto fue ilustrado en el Ejemplo 1, en donde en una primera fase “abierta”, todos los participantes eran conscientes de estar usando un antidepresivo durante 16 semanas. En contraposición, los ensayos clínicos tendrán sujetos “ciegos” cuando ellos desconozcan la asignación. Tradicionalmente, se conocía como “ciego simple” a la condición en que el participante desconocía su asignación; el “doble ciego” incluía además al investigador y el “triple ciego” al evaluador. No obstante, para favorecer la claridad, hoy por hoy se solicita reportar quiénes fueron ciegos a la asignación[16]. Por su parte, el enmascaramiento hace referencia al mismo proceso, pero más bien al hecho de ocultar (“enmascarar”) mediante un “disfraz” el tipo de intervención recibida. Por ejemplo, si la intervención es un fármaco que se administra en un comprimido, los comprimidos de los grupos de intervención (principio activo) y comparación (placebo) tendrán características similares.
Algunos estudios han avanzado incluso más en el mantenimiento del ciego, desarrollando “placebos activos”, es decir, placebos que imitan a la intervención en algunos aspectos: por ejemplo, si un fármaco genera sequedad bucal, los participantes podrían saber si lo están recibiendo o no en función de la presencia de este efecto, por lo que el placebo podría generar también sequedad bucal pero no el efecto relacionado con el principio activo de la intervención[17]. En conclusión, tanto el cegamiento como el enmascaramiento guardan relación con el mismo principio[3],[18],[19],[20],[21]. En el Ejemplo 2 se muestra un ensayo clínico aleatorizado abierto.
Durante el reporte de los resultados suele construirse la llamada “Tabla 1”, en donde se reportan las características biosociodemográficas de interés de cada grupo, tales como sexo, edad, nivel socioeconómico, presencia de alguna patología de interés, uso de algún fármaco, entre otras. Ella tiene un valor descriptivo y analítico ya que permite comparar las diferencias basales entre los grupos.
Clasificación de los ensayos clínicos
Aunque clásicamente se ha relacionado a los ensayos clínicos con el desarrollo de fármacos, este diseño permite la evaluación de cualquier tipo de intervención. La Administración de Alimentos y Medicamentos (FDA, por sus siglas en inglés Food and Drug Administration), agencia reguladora estadounidense, ha presentado una clasificación de los ensayos terapéuticos que evalúan intervenciones farmacológicas, presentada a continuación. En la literatura podrá hallarse una gran mixtura en la clasificación de cada ensayo de acuerdo con esta nomenclatura, la que también se ha usado para ensayos clínicos que analizan intervenciones no farmacológicas[2],[5],[10],[16],[23].
- Estudios preclínicos: detectan problemas de seguridad del producto farmacológico, tales como carcinogenicidad y teratogenicidad. Involucran procesos como síntesis química, pruebas biológicas y estudios toxicológicos. Se realizan en animales.
- Ensayos fase I: estudios farmacológicos sin objetivos terapéuticos, que evalúan toxicidad, parámetros farmacocinéticos y farmacodinámicos, tolerancia, respuesta a distintas dosis y dosis máxima segura. Se realizan en voluntarios sanos y eventualmente en pacientes con enfermedad en estadios avanzados sin otra posibilidad de tratamiento. Por lo tanto, son las primeras pruebas en humanos. Usualmente son estudios abiertos y no controlados que cuentan con menos de 100 participantes.
- Ensayos fase II: corresponden a la primera exploración clínica del tratamiento, en donde debe definirse la posología más adecuada para los estudios fase III. Aportan información preliminar sobre eficacia y seguridad clínica. Se realizan en pacientes portadores de la enfermedad en estudio, pudiendo existir o no un grupo control, pero suelen ser ensayos clínicos aleatorizados. Por lo general incluyen entre 100 y 300 participantes. Se ha descrito una fase II temprana (II a), en donde se realizan estudios piloto para evaluar el perfil de seguridad y actividad de la nueva intervención (mayormente biodisponibilidad), y una fase II tardía (II b), que pretende garantizar aspectos como la seguridad del fármaco y la eventual superioridad de la intervención sobre otra existente. Los estudios fase II pueden actuar como un tamiz de ensayos sobre fármacos que tienen un verdadero potencial para ser evaluados en fase III.
- Ensayos fase III: su objetivo es demostrar el efecto de una intervención en condiciones similares a las que puedan esperarse cuando el fármaco esté disponible para su uso (estudios de confirmación terapéutica). Suelen conducirse en múltiples centros, con muestras mayores a 300 participantes (incluso miles). Se les conoce como “pivotales” o “confirmatorios”, ya que incluyen una estimación del tamaño muestral asociada a una hipótesis estadística a demostrar. Si bien los efectos tóxicos deben haber sido pesquisados en las fases I y II, en fase III se determinarán efectos secundarios frecuentes, indicando el tipo de paciente particularmente susceptible a presentarlos. Los ensayos fase III son el soporte para la autorización del registro y comercialización de un fármaco.
- Ensayos fase IV: evalúan el fármaco en una población distinta a la originalmente estudiada, aportando información adicional sobre riesgos, eventos adversos, beneficios, nuevos usos, efectos a largo plazo (farmacovigilancia), interacciones farmacológicas, entre otros. Se realizan después de la aprobación y comercialización del producto farmacéutico (postmarketing). Los estudios de fase IV corresponden a estudios observacionales (por ejemplo, series de casos, estudios de casos y controles, estudios de cohorte). En consecuencia, están sujetos a los sesgos propios de estos diseños. No obstante, proporcionan información de gran importancia sobre la aplicación del fármaco o intervención en el “mundo real”.
Indistintamente de si los ensayos clínicos estudian intervenciones farmacológicas o no farmacológicas, pueden ser catalogados como unicéntricos en el caso de que se desarrollen por un único grupo de investigación en solo un centro hospitalario; o multicéntricos, cuando un protocolo de investigación en común es ejecutado por más de un grupo de investigación en más de un centro. Estos últimos permiten estudiar a un mayor número de participantes en menos tiempo, con conclusiones más fiables y representativas de la población. Sin embargo, su planificación, coordinación, supervisión, gestión y análisis de datos es más compleja[16],[23].
Finalmente, es frecuente encontrar en la literatura publicada la denominación de “estudio piloto” a ciertos ensayos clínicos (Ejemplo 3)[24]. Estos corresponden a ensayos preliminares cuyo objetivo es realizar un sondeo para ejecutar un ensayo clínico posterior de mayor relevancia. Los estudios piloto entregan luces respecto a la precisión de la hipótesis, definición de la muestra (criterios de elegibilidad) y de la intervención, estimación del tiempo requerido para el estudio e información sobre los eventuales datos perdidos. Además, de manera muy importante, aportan evidencia para la determinación del tamaño muestral del ensayo clínico posterior[16],[25].
Aspectos bioéticos y registro de protocolo
La Declaración de Helsinki fue desarrollada por la Asociación Médica Mundial en 1964 para ser una guía ética de la investigación con seres humanos, incluyendo aspectos como deberes de quienes investigan en seres humanos, importancia del protocolo de investigación, investigación con personas vulnerables, consideraciones sobre riesgos y beneficios, relevancia del consentimiento informado, mantenimiento de la confidencialidad e información de los hallazgos a los participantes del estudio. Aunque no es legalmente vinculante en sí misma, muchos de sus presupuestos están contenidos en las legislaciones de los países al respecto, por lo que debe considerarse en la construcción de cualquier estudio con seres humanos. La declaración cuenta con múltiples revisiones a la fecha[17],[26] (https://www.wma.net/what-we-do/medical-ethics/declaration-of-helsinki/).
Para dar inicio a un ensayo clínico, es necesario tener en mente el principio básico estipulado por la Declaración de Helsinki[26] en cuanto a las posibles intervenciones a estudiar: los posibles beneficios, riesgos, costos y eficacia de toda intervención nueva deben ser evaluados mediante su comparación con las mejores intervenciones probadas, excepto en las siguientes circunstancias:
- El uso de placebo o no tratamiento es aceptable en estudios donde no exista una intervención probada.
- Cuando por razones metodológicas científicamente sólidas y convincentes, sea necesario para determinar la eficacia y la seguridad de una intervención el uso de cualquier intervención menos eficaz que la mejor probada, el uso de un placebo o ninguna intervención.
Los ensayos clínicos requieren de un protocolo de investigación que debe ser evaluado y aprobado por un comité ético-científico (así como toda investigación con seres humanos[1],[27],[28]), y registrado en algún repositorio disponible antes de que ocurra la inscripción del primer participante[29],[30]. Su finalidad es detectar cualquier desviación del protocolo original una vez realizado el estudio, garantizando que los autores reporten los desenlaces que declararon en un comienzo como clínicamente relevantes, lo que evita, por lo tanto, el informe selectivo de resultados[31]. Este proceso otorga transparencia y visibilidad a la investigación clínica, permitiendo a quienes desarrollen futuros ensayos clínicos y revisiones sistemáticas de ensayos clínicos, contar con un mapa general de la investigación en curso. Todo esto se ha conceptualizado en el modelo de Buenas Prácticas Clínicas. Se trata de un estándar para el diseño, conducción, desempeño, monitoreo, auditoría, registro, análisis y reporte de ensayos clínicos, que cautela la confiabilidad de los resultados en un marco de integridad investigativa y confidencialidad de los participantes[10].
A fines del siglo XX, se originaron diversos registros públicos para ensayos clínicos. En Estados Unidos se creó el registro Clinical Trials, financiado con fondos públicos (http://clinicaltrials.gov), mientras que en Europa se estableció el registro ISRCTN (http://isrctn.com), reconocido por la Organización Mundial de la Salud y por el Comité Internacional de Editores de Revistas Médicas (ICMJE, por sus siglas en inglés International Committee of Medical Journal Editors) y respaldado por el Medical Research Council y el Programa de Investigación y Desarrollo del Sistema Nacional de Salud, ambas organizaciones británicas[32]. Por su parte, Cochrane dispone del Cochrane Central Register of Controlled Trials (https://www.cochranelibrary.com/central) y la Organización Mundial de la Salud cuenta con la Plataforma Internacional de Registro de Ensayos Clínicos (ICTRP, por sus siglas en inglés International Clinical Trials Registry Platform) (https://www.who.int/ictrp/en/)
Componentes y procedimientos esenciales en los ensayos clínicos
Reclutamiento y aleatorización de participantes
El reclutamiento de participantes para un ensayo clínico es por lo general un muestreo no probabilístico, donde se incorporan aquellos sujetos que cumplen con los criterios de elegibilidad establecidos en el protocolo del estudio. También se le conoce como “muestreo por conveniencia”. Luego, los sujetos son aleatorizados a una intervención o a su comparación, lo que, como se abordará más adelante, es el mejor método para controlar los sesgos de selección y confusión[3],[33],[34]. Esta asignación no discrecional de participantes a los grupos en estudio debe realizarse estrictamente por azar para garantizar que todos los participantes tienen la misma probabilidad de ser incluidos a cualquiera de los grupos. A medida que avanza este proceso, los grupos tienden a ser más homogéneos, tanto en las variables de confusión como en otras variables que podrían afectar sobre el desenlace, pero que no podrían ser evaluadas a priori.
La aleatorización puede realizarse mediante una tabla de números aleatorios hallada en un libro de estadística, pero usualmente se emplean métodos informáticos de aleatorización, como una secuencia generada por un computador. Una mención particular merece el ocultamiento de la secuencia de aleatorización, la que debe ser desconocida por los investigadores y participantes del ensayo clínico, de modo de no poder predecir el grupo al que será asignado el siguiente participante incluido.
La estimación de la cantidad de participantes que deben ser aleatorizados (cálculo del tamaño de la muestra) es parte primordial de la aleatorización. ¿Cuántos participantes se requieren para igualar los factores de confusión entre los grupos de intervención? No necesariamente un número mayor será mejor, ya que podría exponerse innecesariamente a personas a los riesgos de una intervención. De este modo, si la cantidad de pacientes aleatorizados es menor que el tamaño de la muestra estimada, sus resultados estarán sesgados aun cuando haya existido la aleatorización[8].
Entre los distintos tipos de aleatorización se encuentran la aleatorización simple, basada en una secuencia única de asignaciones aleatorias, manteniendo completamente la asignación al azar. En ensayos clínicos con grandes tamaños de muestra, la aleatorización simple puede generar números similares de participantes entre los grupos, pero en estudios que involucran pocos participantes puede resultar en cantidades desiguales en cada grupo[35]. Otra forma de aleatorización es la aleatorización en bloques, cuyo objetivo es asegurar que los tamaños de cada grupo sean similares[17]. Cada bloque contiene a un número similar de participantes asignado a cada tratamiento, donde el total de participantes ha sido predeterminado por los investigadores. Luego, los bloques son asignados aleatoriamente a cada grupo.
El problema de la aleatorización en bloques es que los grupos generados pueden ser desiguales en cuanto a ciertas variables de interés[35]. Ante esto, la aleatorización estratificada se aplica para garantizar que se asignará a cada grupo una cantidad similar de participantes con una característica de importancia para el estudio, las que deben identificarse por los investigadores. En este tipo de aleatorización, se configuran distintos bloques de participantes con combinaciones de covariables que puedan influir en la variable dependiente que se quiere explicar (aleatorización según factores pronósticos). Luego, se realiza una aleatorización simple dentro de cada estrato para asignar a los sujetos a alguno de los grupos de intervención. Por lo tanto, se puede intuir que para llevar a cabo una aleatorización estratificada, es necesario conocer con exactitud las características de cada sujeto[17],[35],[36] (Ejemplo 4).
Finalmente, un método de aleatorización que se ha empleado en los ensayos clínicos con un tamaño de muestra pequeño es la aleatorización adaptativa, ya que en ella se asigna secuencialmente un nuevo participante a un grupo de intervención en particular, considerando las asignaciones previas de los participantes además de las covariables específicas. La aleatorización adaptativa usa el método de minimización, evaluando el desbalance del tamaño muestral entre múltiples covariables, lo que podría ocurrir al aplicar una aleatorización simple en un ensayo clínico con una muestra reducida[35],[37],[38].
Cualquier asignación que no responda al azar, tal como la asignación por alternancia (por ejemplo, el primer participante es asignado al grupo intervención, el segundo al grupo control y así sucesivamente), de acuerdo al día de la semana, según la letra inicial del primer apellido, entre otros, no puede calificarse como una asignación aleatoria[4].
Medición de resultados
Idealmente, deben aplicarse métodos de enmascaramiento que aseguren el ciego de los participantes, como fue previamente señalado. El ciego permite que el énfasis que pongan los encargados del ensayo en la medición de resultados será el mismo para todos los grupos y que los pacientes no tendrán la influencia de saber si están en el grupo intervención o no, lo que disminuiría las respuestas subjetivas al tratamiento. Por tanto, el ciego previene la ocurrencia del sesgo de medición.
Tres fenómenos relacionados que pueden suceder a nivel de los participantes son los siguientes: el efecto placebo[40],[41], el efecto nocebo[40] y el efecto Hawthorne[42],[43]. El primero se asocia a una mejoría reportada por el participante después de recibir una sustancia sin desenlaces relevantes demostrados, esto es, un placebo. Por el contrario, en el efecto nocebo, una sustancia o intervención sin efectos médicos empeora el estado de salud de una persona debido a las creencias negativas que el participante tiene sobre ella. El efecto Hawthorne, también conocido como “efecto del observador”, ocurre cuando los participantes de un ensayo clínico modifican su conducta habitual al saber que están siendo observados por un tercero, lo que impacta en los efectos de la intervención. Estos tres fenómenos son más prominentes en ensayos clínicos aleatorizados que analizan desenlaces reportados por los participantes, es decir, donde existe un componente subjetivo mayor (Ejemplo 5).
Análisis de datos
Una vez medidos los desenlaces de interés tras el tiempo necesario, es esencial el poder incluir en el análisis estadístico primario a todos (o la mayor parte de) los participantes aleatorizados, ya que por algún motivo, algunos sujetos podrían abandonar el tratamiento, lo que debe ser evitado por los investigadores. El análisis primario debe realizarse incluyendo y analizando a todos los pacientes como parte del grupo original al que fueron asignados, lo que se conoce como análisis por intención de tratar. El principio del análisis por intención de tratar es mantener hasta el final del estudio el objetivo logrado con la aleatorización, es decir, el balance de los factores pronósticos conocidos y desconocidos y de los factores de confusión, disminuyendo la probabilidad de sesgar los resultados[45].
No obstante, este enfoque se ve dificultado cuando existe una baja adherencia al tratamiento asignado, o cuando el ensayo no logra el seguimiento de todos los pacientes. Algunos investigadores resuelven este problema excluyendo del análisis a los pacientes que no adhirieron al protocolo, lo que se conoce como análisis por protocolo. Este solo incorpora a participantes que recibieron la intervención asignada por la aleatorización, lo que en un comienzo puede parecer razonable, pues interesa conocer los efectos de la intervención en los pacientes que realmente la recibieron. Sin embargo, es reconocido que la adherencia habitual a cualquier tratamiento suele ser inferior a la estimada teóricamente, por lo que el análisis por protocolo no representaría lo que sucede en la realidad[46]. A su vez, se ha demostrado que los pacientes menos adherentes a una terapia, aunque esta sea un placebo, tienden a tener peor pronóstico que los que sí adhieren a ella[47]. Al excluirlos del análisis, se está estudiando los pacientes de mejor pronóstico, efecto que se magnifica si se decidiera incluir al grupo de participantes que no recibió la intervención (y que podrían considerarse como semejantes al grupo control) en el grupo control, perdiendo así el efecto de la aleatorización[48] (Ejemplo 6).
Tipos de estudios experimentales en seres humanos
En este artículo se han abordado mayormente los conceptos relacionados con el ensayo clínico aleatorizado clásico, cuyas variantes y especificaciones se describen a continuación. No obstante, existen otros diseños que también estudian los efectos de una intervención sobre los participantes.
Ensayo clínico aleatorizado clásico
Corresponde al ensayo fase III de la clasificación anterior. Se aleatorizan los participantes a recibir una intervención de prueba o una comparación. La aleatorización garantizaría que las características de los participantes se distribuyan homogéneamente entre los grupos, por lo tanto, cualquier diferencia significativa en el desenlace entre los grupos puede ser atribuida a la intervención y no a otro factor no identificado. Si el ensayo clínico busca evaluar una intervención en condiciones ideales (no cotidianas) y controladas de manera rigurosa, se trata de un ensayo clínico explicativo (estudios de eficacia). Mientras que, si su evaluación se da en un contexto que emula las circunstancias de la vida o la práctica clínica cotidiana, recibe el nombre de ensayo clínico pragmático[49]. En este sentido, se han desarrollado herramientas para evaluar el nivel de pragmatismo de un ensayo clínico[50].
Otra denominación se relaciona con el objetivo del estudio, ya que existen ensayos clínicos aleatorizados cuyo objetivo es demostrar la superioridad, equivalencia o no inferioridad de una intervención sobre otra[51]. En el primer caso, se busca probar que un tratamiento es mejor que otro. En el segundo, será apropiado conducir un estudio de equivalencia si el nuevo tratamiento tuviera similar eficacia, pero ciertos beneficios como menos efectos adversos, un uso más simple o un menor costo económico. Los estudios de no inferioridad pueden considerarse como un caso especial de los estudios de equivalencia. Estos solo analizan que una intervención no es peor que otra ya existente[17].
En virtud del diseño del estudio encontraremos el ensayo clínico aleatorizado paralelo. Es el modelo más frecuente, en donde cada grupo de participantes recibe una intervención simultáneamente. Por su parte, en los ensayos clínicos aleatorizados cruzados (crossover), cada participante recibe consecutivamente cada intervención en estudio, de modo de que cada sujeto es su propio comparador. Por lo tanto, requieren de un tamaño de muestra menor, ya que los sujetos aleatorizados inicialmente al grupo de intervención luego recibirán la comparación y viceversa, por lo que sumarán participantes a ambos grupos. Una desventaja de este diseño es el fenómeno de carryover, en donde los efectos de la primera intervención pueden interferir con los efectos de la segunda. Por ende, este tipo de ensayos es útil en intervenciones que duran un periodo corto. De todos modos, es conveniente espaciar temporalmente ambas intervenciones (periodo de lavado), en orden a disminuir la probabilidad de que la primera interfiera con la segunda[52],[53]. Por lo tanto, al suspender la intervención, la condición del sujeto que la recibió debe ser la misma que antes de recibirla; ya que si cambia, la segunda intervención se aplicaría sobre un participante distinto al que recibió la primera. Es por esta razón que este diseño se limita al estudio de condiciones crónicas y estables en el tiempo[54] (Ejemplo 7).
Otro diseño existente es el ensayo clínico factorial, el que permite responder conjuntamente a dos o más preguntas de investigación. Un ejemplo es cuando dos o más intervenciones son evaluadas separadamente y en combinación contra un control. Los participantes son aleatorizados dos o más veces a alguno de los grupos de intervención, dependiendo del número de terapias a estudiar. La gran ventaja es que aporta más información respecto a un estudio con diseño en paralelo. Además, permitiría evaluar la interacción entre dos tratamientos[56].
Otra modalidad de ensayo clínico aleatorizado es aquella en que se aleatoriza por conglomerados (clusters). Vale decir, se asignan aleatoriamente grupos de participantes (por ejemplo, centros de salud, áreas geográficas) a una intervención o su comparación. Son útiles en el estudio de individuos con características biológicas o psicosociales similares y cuando la intervención analizada tiene un efecto grupal, tales como intervenciones no farmacológicas en políticas públicas comunitarias[57],[58] o el efecto de una vacuna[5].
Finalmente, existen ensayos clínicos aleatorizados de descontinuación. En ellos, los pacientes que ya reciben algún tratamiento son aleatorizados a continuar con su terapia o descontinuarla y recibir un placebo. Los diseños de descontinuación se aplican en terapias crónicas no curativas cuyo efecto es de pequeña magnitud[54].
Estudios cuasi-experimentales
Se caracterizan por no presentar un proceso de aleatorización de los participantes al grupo intervención o al grupo comparador, por lo que también se conocen como ensayos controlados no aleatorizados. Si bien existen numerosos ejemplos de tipos de estudios cuasiexperimentales en la investigación biomédica, este diseño proviene del campo de la psicología y las ciencias sociales[59], donde se ha considerado que en ciertas circunstancias no es posible la asignación aleatoria de sujetos a condiciones experimentales. Existen diversos tipos de estudios cuasiexperimentales; entre ellos, los diseños antes/después o pretest/postest y las series de tiempo interrumpidas. Destacaremos el diseño de antes-después, en el cual se mide una misma variable de manera previa y posterior a una intervención (cada participante actúa como su propio control). Sus resultados son útiles cuando el efecto es de gran magnitud, se observa consistentemente en la mayoría de los participantes y, por ende, es improbable que se explique por efecto del azar[4] (Ejemplo 8).
Los estudios cuasiexperimentales suelen ser más simples e implican un menor costo económico que los ensayos clínicos aleatorizados. Es por esto que constituyen una opción cuando la asignación aleatoria es poco factible de realizar, cuando existe un impedimento bioético o cuando se requiere realizar la intervención en condiciones naturales. Sus desventajas se asocian con una gran susceptibilidad a los sesgos de confusión y selección. Además, el efecto placebo y el efecto Howthorne son especialmente relevantes, lo que podría aminorarse si es que los sujetos no son conscientes de la intervención de la que participan[60],[61]. Durante el análisis de los datos deben considerarse idealmente métodos estadísticos para grupos pareados, ya que se realizarán mediciones repetidas en el tiempo a un mismo grupo de sujetos[62]. Por todo lo anterior, la interpretación y extrapolación de los datos obtenidos desde los estudios no aleatorizados es compleja[17].
Estudios experimentales con controles externos o históricos
Utilizan como grupo control a sujetos que no provienen de la misma población desde la que se obtuvo la muestra (muestra no concurrente o no contemporánea). Por lo tanto, la comparación se realizará a partir de datos de pacientes ya publicados o desde registros de una institución de salud, es decir, con personas que ya han recibido un tratamiento y una evaluación. Una desventaja de esta metodología son las diferencias que puedan existir entre el grupo intervención y el grupo control, haciéndolos poco comparables[4], ya que existen diferencias en el contexto temporal en que se aplicaron los tratamientos, las personas que realizaron la intervención, entre otros[17].
Medidas de asociación
Debido a que se trata de un diseño prospectivo, la medida de asociación a usar será el riesgo relativo (RR), el que se entiende como la razón de los riesgos absolutos entre el grupo de individuos expuestos a la intervención y los no expuestos. Si el riesgo relativo es igual a 1, se asume que no existe asociación entre la intervención y el desenlace (esto es, el intervalo de confianza asociado no incorpora el valor 1). Si es mayor a 1, la intervención aumenta la probabilidad de que suceda el desenlace; mientras que, si es menor a 1, la intervención disminuye la probabilidad de ocurrencia del desenlace. La interpretación será distinta en virtud del desenlace medido, ya que este puede ser favorable o desfavorable. Por lo tanto, si el resultado estudiado es disminución de ánimo depresivo, un riesgo relativo mayor a uno será favorable, mientras que si se mide mortalidad, un riesgo relativo mayor a uno será desfavorable[63],[64],[65].
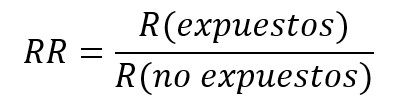
Otro modo para expresar la magnitud de asociación es mediante diferencias. Una de ellas es la reducción absoluta de riesgo (RAR), riesgo atribuible o reducción de riesgo, que corresponde a la diferencia entre los riesgos del grupo control (no expuesto) e intervención (expuesto). Esto quiere decir que la reducción de riesgo es atribuida a la intervención. Si su resultado es negativo, se interpreta como un aumento absoluto del riesgo[63],[64],[65].
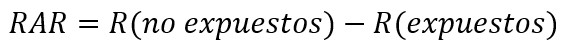
Por su parte, la reducción relativa de riesgo (RRR) corresponde a la diferencia de riesgo entre los dos grupos respecto al grupo control, o cociente entre la reducción absoluta de riesgo y el riesgo del grupo control. Si su resultado es negativo, se interpreta como un aumento relativo del riesgo. La reducción relativa del riesgo frecuentemente sobreestima los efectos del tratamiento, por lo que la reducción absoluta de riesgo debería reportarse cuando sea posible[63],[64],[65].
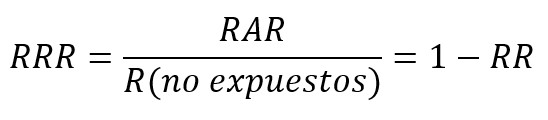
Una medida derivada de la reducción absoluta de riesgo es el número necesario a tratar (NNT), la que cuantifica el número de pacientes a tratar para evitar que ocurra un evento[63],[64],[65]. Por ejemplo, si el número necesario a tratar para un antibiótico fuese de 15, se interpreta que se debe tratar a 15 pacientes para la recuperación de un cuadro infeccioso.
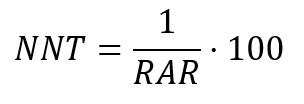
Por otra parte, el número necesario a dañar corresponde a un índice de los eventos adversos asociados a un tratamiento, significando el número de pacientes que debería recibir un tratamiento en lugar de otro para que un paciente adicional presente un evento perjudicial. Cuando el resultado del número necesario a tratar es negativo, debe interpretarse como el número necesario a dañar[63],[64],[65].
Si bien la reducción absoluta del riesgo, la reducción relativa del riesgo y el número necesario a tratar otorgan información sobre la magnitud del efecto del tratamiento, están fuertemente relacionadas a la variabilidad del parámetro medido y al tamaño de la muestra. Por ello, deben reportarse junto a intervalos de confianza que entreguen información sobre la precisión de los hallazgos[66].
Sesgos
Un sesgo presente en cualquier tipo de investigación es el sesgo de confusión, ya que nunca podrá eliminarse del todo. Sin embargo, los ensayos clínicos logran reducirlo significativamente gracias al proceso de aleatorización, el que permite la distribución homogénea de las variables conocidas y desconocidas entre los grupos de estudio. A continuación, se nombran algunos de los sesgos observados en los ensayos clínicos.
Sesgo de selección
Ocurre cuando los métodos para seleccionar la muestra desde la población favorecen a un grupo respecto al otro. También aparece cuando no se incluye a alguna proporción relevante de la población objetivo. Este sesgo es controlado con una adecuado proceso de selección de participantes y con su posterior aleatorización[67].
Sesgo de desarrollo (performance bias)
Existe una diferencia sistemática entre grupos respecto al cuidado y seguimiento entregado. Por ejemplo, está presente cuando los investigadores llevan un seguimiento más estricto de los pacientes asignados a la intervención bajo estudio. La estandarización de los procedimientos, el entrenamiento adecuado del personal y el enmascaramiento combaten esta fuente de error sistemático[67],[68].
Sesgo de detección o información
Sucede cuando el desenlace en estudio es “detectado” diferencialmente entre los grupos, lo que puede conducir a resultados diferentes. El sesgo de detección es más prominente durante el registro de desenlaces subjetivos reportados por los participantes (por ejemplo, respuesta analgésica). De esta forma, si un investigador registra los resultados observados de un modo que avale su creencia, emergerá el sesgo de detección. Este sesgo se controla mediante el enmascaramiento[67],[68].
Sesgo de desgaste o post aleatorización (attrition bias)
Se presenta cuando existen diferencias sistemáticas en el seguimiento de los participantes del ensayo clínico, ocasionando por ejemplo pérdidas de seguimiento, las que incrementan la incertidumbre en los resultados. Este sesgo se controla realizando un análisis por intención de tratar[67],[68].
Sesgo de reporte (reporting bias)
Este sesgo se pesquisa durante la presentación de los resultados, donde puede ocurrir un reporte selectivo de los desenlaces de mayor interés o de aquellos que demuestran la hipótesis en estudio, lo que aumentaría el impacto del estudio y la probabilidad de que sea publicado. El sesgo de reporte puede evaluarse mediante la revisión del registro inicial del ensayo clínico y/o de su protocolo publicado para luego compararlo con lo que se ha reportado[67],[68].
Pautas de reporte
El adecuado reporte de los ensayos clínicos es de gran importancia, pues permite comprender e interpretar los resultados y las conclusiones, así como garantizar su reproducibilidad[69]. Con el fin de estandarizar criterios en el reporte de los ensayos clínicos y facilitar la lectura crítica y la interpretación, aparece a mediados de los noventa la iniciativa Consolidated Standards of Reporting Trials (CONSORT, por sus siglas en inglés)[70], la que está en constante revisión, actualización y especialización. Esta propuesta está constituida por 25 ítems agrupados en seis dominios: título y resumen, introducción, metodología, resultados, discusión y otra información, debiendo agregarse además un diagrama de flujo de los participantes en el ensayo, lo que permite una rápida comprensión de los aspectos más relevantes de la ejecución del estudio. Existen múltiples versiones de Consolidated Standards of Reporting Trials adaptadas para intervenciones en específico: para estudios de no inferioridad[71], para estudios piloto[72], para estudios con intervenciones herbales[73], para ensayos clínicos pragmáticos[74], para ensayos clínicos con intervenciones psicológicas y sociales[75], para ensayos clínicos de participante único[76], para ensayos clínicos cruzados[53], entre otras. Brevemente, en la lectura de un ensayo clínico publicado deben responderse las siguientes preguntas[5]:
- ¿Se trata de un ensayo clínico de alta calidad que aborda una pregunta importante?
- ¿La aleatorización se realizó adecuadamente?
- ¿Cuán completo fue el seguimiento? ¿Fue similar entre los grupos del estudio?
- ¿Se buscaron desenlaces positivos y negativos? ¿Fueron ciegas esas evaluaciones?
- ¿Los resultados se aplican a la práctica clínica?
- ¿Los pacientes fueron adecuadamente descritos?
- ¿La intervención fue apropiadamente descrita?
- ¿Se realizó un análisis por intención de tratar o por protocolo?
Por otra parte, la iniciativa Transparent Reporting of Evaluations with Nonrandomized Designs (TREND, por sus siglas en inglés)[77], está destinada al reporte de estudios de intervención no aleatorizados. Consta de 22 ítems agrupados en cinco dominios: título y resumen, introducción, métodos, resultados y discusión.
Paralelamente, para evaluar la calidad de los ensayos clínicos se han presentado escalas como la de Jadad[78] y la desarrollada por Cochrane[79].
Consideraciones finales
Usualmente, la primera tabla de resultados que se reporta en las publicaciones de ensayos clínicos es la “Tabla 1”, donde se describen condiciones de interés de los participantes. Es igualmente frecuente encontrar en la literatura publicada que esta “Tabla 1” presente los valores p resultantes de la comparación con una prueba estadística de las características entre el grupo de intervención y el grupo comparador, siendo esperable que estos valores p sean mayores al nivel de significancia estadística. Ello demostraría que los grupos no difieren en sus características biosociodemográficas y, por lo tanto, ellas no se comportarían como variables de confusión.
Sin embargo, esto no es metodológicamente correcto, ya que la hipótesis estadística subyacente al ensayo clínico y asociada a la pregunta de investigación se relaciona con la estimación de una diferencia entre dos intervenciones y no con la evaluación de la frecuencia de una variable entre grupos. Este hecho ha sido enfatizado por la guía Consolidated Standards of Reporting Trials[70],[80]. Algunos autores han dispuesto una solución: si existe una diferencia mayor al 10% entre la frecuencia de las variables estudiadas, podrá suponerse que existe un potencial efecto de confusión. Si esta diferencia existe, quiere decir que la aleatorización no controló todo el efecto de la confusión, por lo que los resultados deben evaluarse teniendo en mente este efecto residual o, mejor aun, realizar un modelo de regresión multivariado que incorpore a la variable en cuestión para evaluar su efecto como factor de confusión[8].
El carácter prospectivo de los ensayos clínicos permite encontrar relaciones causales y adoptar medidas para asegurar la calidad de los datos obtenidos. No obstante, es necesario que los investigadores esperen el tiempo suficiente (a veces bastante tiempo) para que los desenlaces de interés ocurran. En este punto vale la pena reconocer el análisis de supervivencia, método estadístico mayormente usado en los estudios de cohorte[28], que permite analizar el tiempo en que ocurre un desenlace esperado y comparar las curvas temporales entre distintos grupos. Otro método estadístico aplicado en los ensayos clínicos es el análisis secuencial, el que consiste en la realización de análisis intermedios para evaluar la necesidad de continuar o detener un ensayo en virtud de la demostración de la hipótesis y el balance costo-beneficio, obedeciendo a ciertas reglas de detención. El análisis secuencial debe especificarse en el protocolo del estudio[81],[82].
Algunas críticas que se han hecho a los ensayos clínicos aleatorizados se relacionan con la poca representatividad que puede tener la muestra de participantes respecto al real espectro en la práctica clínica, cuyo muestreo es no probabilístico y se realiza sobre personas seleccionadas con criterios de elegibilidad rigurosos. Asimismo, la estandarización de la intervención tampoco se asemeja a lo que sucede realmente, en donde las intervenciones serían menos controladas y más heterogéneas. Por esto, la validez externa (extrapolación de resultados) debe evaluarse con cautela, ya que los resultados del ensayo clínico pueden distar de lo observado en la práctica clínica, en donde intervienen múltiples factores que no fueron controlados en el estudio[2].
No todas las interrogantes pueden ser respondidas mediante un estudio de experimental, como por ejemplo, el análisis de los factores de riesgo para el desarrollo de neoplasia pulmonar, donde no sería ético que un grupo de personas recibieran una intervención deletérea para su salud. En estos casos cobran importancia los estudios observacionales[28]. El estudio de los eventos adversos asociados al uso de fármacos debe reservarse para estudios observacionales o estudios experimentales de fases tempranas, pero no en experimentos clínicos con seres humanos. De esta forma, el análisis de eventos adversos de baja ocurrencia permanece como un desafío, puesto que los estudios disponibles cuentan en general con una baja potencia (vinculada al bajo tamaño de muestra), se requieren grupos de comparación muy similares y el análisis de la relación causa-efecto es complejo[2].
Es necesario diferenciar los desenlaces clínicamente relevantes o significativos de los desenlaces subrogados, intermedios, indirectos o sustitutos[83]. Los desenlaces clínicamente relevantes orientan respecto al uso de una intervención y dan luces sobre el efecto de la terapia, es decir, la medida directa de cómo un paciente se siente, vive y funciona. Por su parte, los desenlaces subrogados pueden corresponder a un parámetro de laboratorio o un signo físico que no miden directamente el beneficio clínico central de la intervención. Así, los primeros se asocian a la persona en sí misma, mientras que los segundos a la fisiopatología de la enfermedad. Esta consideración es importante al leer ensayos clínicos aleatorizados, pues podría ocurrir que un estudio reporte múltiples desenlaces subrogados positivos pero no resultados clínicamente relevantes.
Finalmente, aunque los ensayos clínicos aleatorizados son la piedra angular para estudiar la eficacia y seguridad de una terapia, una revisión sistemática que metaanalice los resultados de múltiples ensayos clínicos que estudiaron una misma intervención, reportará un nivel de evidencia aun mayor, ya que ofrece un estimador combinado del efecto de todos los estudios primarios que incluyó[84]. En cualquier sentido, los resultados obtenidos por un ensayo clínico deben publicarse, de lo contrario, se contribuiría al sesgo de publicación al publicarse solo los resultados que son positivos a la hipótesis establecida en el protocolo. Esto favorece que la comunidad científica observe un efecto magnificado de una intervención[6], lo que impacta negativamente en la sociedad. Se trata de un hecho contraproducente con el bien interno de la investigación, esto es, el bien social.
 Tamaño completo
Tamaño completo 